
|
|
|
Only the colored codons/residues are allowed at the N-terminus of a nascent protein sequence, as it emerges from a ribosomal exit tunnel. A blue residue at the N-terminus is normally N-acetylated, a green residue may be N-acetylated (but often isn't) & an orange residue is not N-acetylated. Exception: any protein with a sequence that starts with "XP" is not N-acetylated for X = S, T, A, C, V or G. 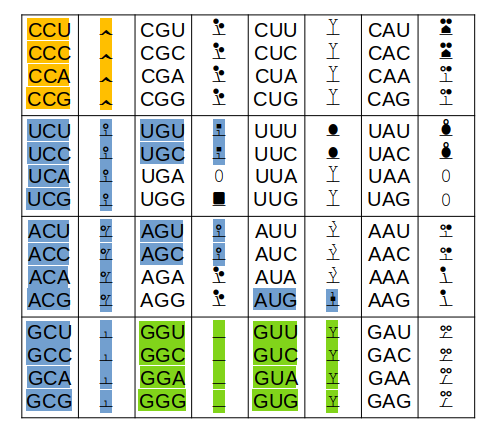
Other exceptions: 1 MR... will not be N-acetylated (tubulin exception) 1 acetyl-MDDD (actin B exception) will mature to 1 acetyl-MEEE (actin G1 exception) will mature to The actin PTMs occur in the cytoplasm via enzymes dedicated to the purpose.
Something that has shaped the practice of proteomics was to base
as much as possible on sets of rules rather than statistics or other types
of evidence derived from the data itself.
The most fundamental of
these rules are the "canons" (fancy rules) used to select a single splice
variant to stand in for all of the possible translations of a gene. A particularly
influential set of 5 rules for this choice is used by UniProt
(see here).
It should come as no surprise that these rather trivial rules regularly choose a
splice that isn't very representative of what is found in biological samples, which can cause
bias and systematic interpretation errors in the results of typical data interpretation methods.
The fumarase gene is responsible for both a mitochondrial & cytoplasmic form that differ only in that the cytoplasmic form has an N-terminal acetylation. When the translation of FH:r initiates at its first Met codon ... ACC AUG UAC ... the sequence has a 44 residue mitochondrial transit peptide that is removed on the passage of the sequence into the mitochondrial inner matrix. Human fumarate hydratase (FH:p) 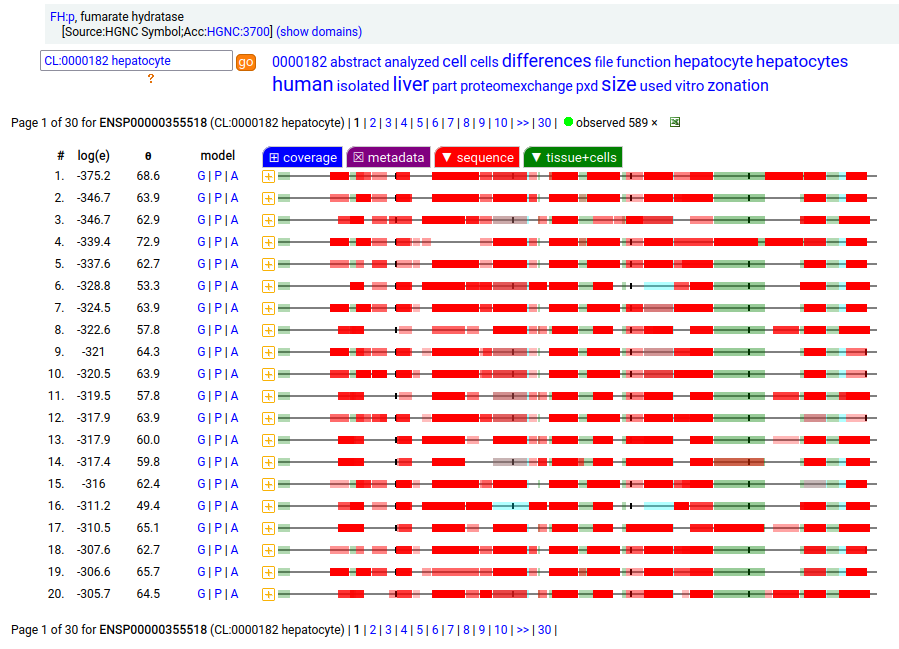
It seems that about 10% translations skip this first Met-codon & instead initiate at the second one ... CGA AUG GCA ... resulting in a protein with the N-terminal tryptic peptides 45 ac-ASQNSFR 51 lacking the transit peptide, so it stays in the cytoplasm. The gene pulls off this trick by having the second Met codon as the last codon of the first exon, which also codes for the entire transit peptide. 
MTCH1:p is interesting: it has 2 initiation variants that are observed to be tissue-dependent. Brain tissue has the M1 initiating variant as the dominant form of this membrane protein, while liver tissue has the M19 initiating variant as the dominant form. Human mitochondrial carrier 1 (MTCH1:p) proteomics canon_folly 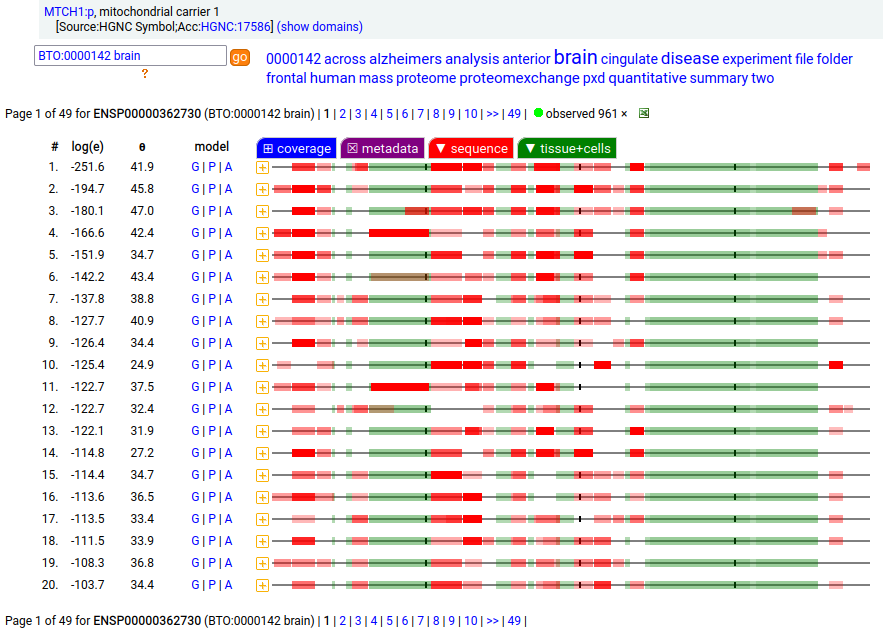 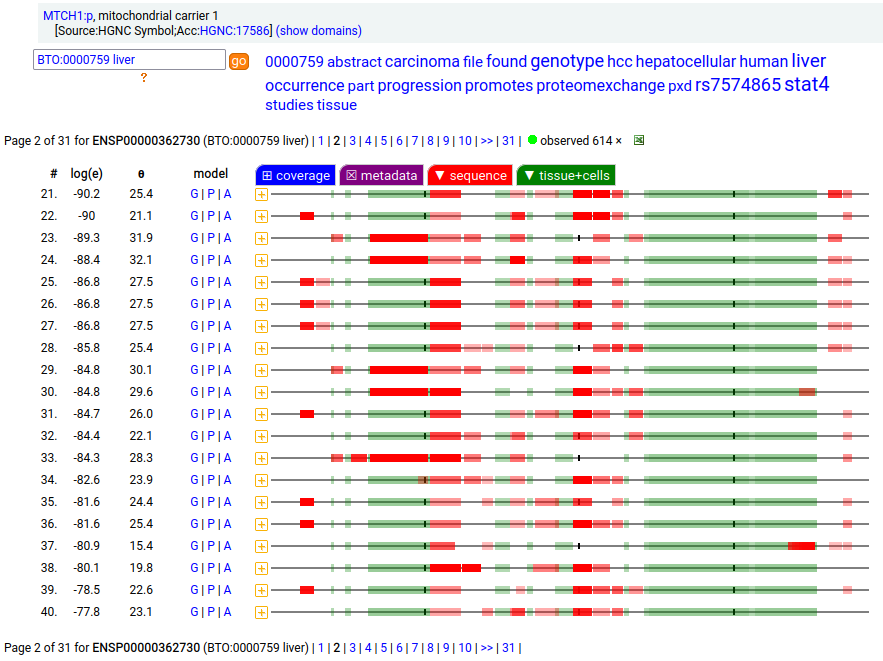
This sequence also has overlapping tryptic peptides that can be used to monitor the amount of each variant present in a sample. Mouse appears to use the same mechanism & shares the same tissue variant distribution. 
MTCH2:r has an AUG at about the same position in its mRNA sequence as MTCH1:r, but it does not generate observable levels of an initiation variant protein product. The coverage diagram for the canonical (252 aa) form of DUT:p may look like there is a splice selection error, but there is something more subtle going on. In fact, this diagram is characteristic of the removal of a longish mitochrondrial targeting peptide (1-69,70), resulting in a shorter mature enzyme in the mitochondrial matrix with a slightly ragged N-terminus. Human deoxyuridine triphosphatase (DUT:p) 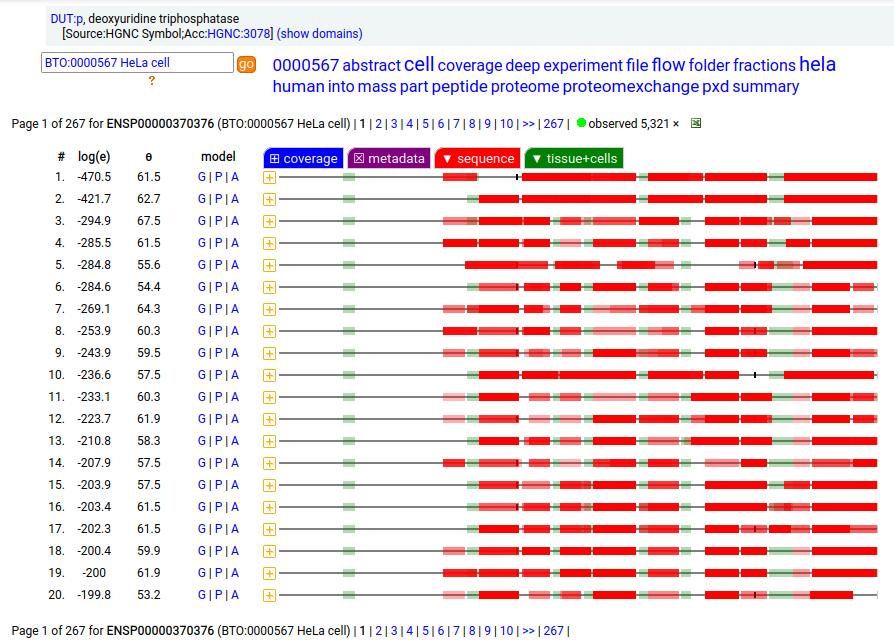
But, DUT:p is needed in both the mitochondria & cytoplasm. Rather than having a 2nd gene, in this case DUT:p has another splice variant (162 aa) that effectively removes the targeting peptide but retains the enzymatic portion of the sequence by removing the 2 exons coding for the peptide in the longer splice & substituting a shorter exon with an AUG in the right place. 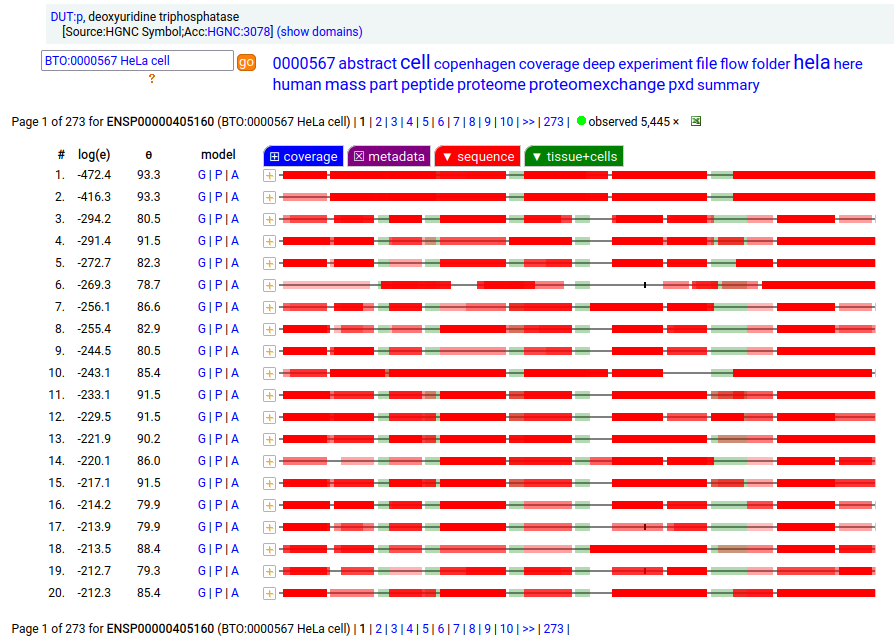
These two splices can be distinguished in proteomics data by examining the mature form N-terminal tryptic peptides corresponding to the two different protein sequences. Note: N-terminal proline residues do not undergo co-translationally acetylation. 
GARS1:p is another case where the coverage diagram suggests there may be some RNA high jinks at the N-terminus. The full length (739 aa) translation has a mitochondrial transit peptide (1-35,37,39) that is removed during import into the mitochondrial matrix, leaving the enzyme with a ragged N-terminus. Human glycyl-tRNA synthetase 1 (GARS1:p) 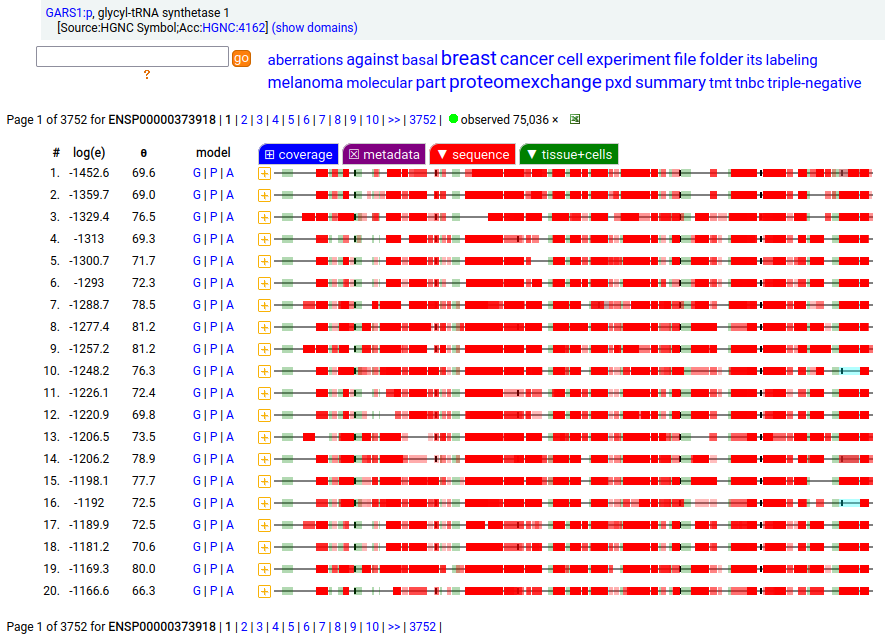
To create the more abundant cytoplasmic protein, neither a new gene nor splice is necessary. Instead, it is generated by simply ignoring M1 during translation, skipping the transit peptide and initiating at M55 (685 aa). While any tryptic peptide with a residues in the region (35-54) must be from the mitochondrial enzyme, only peptides with acetylated M55 at their N-terminus are unambiguously cytoplasmic. 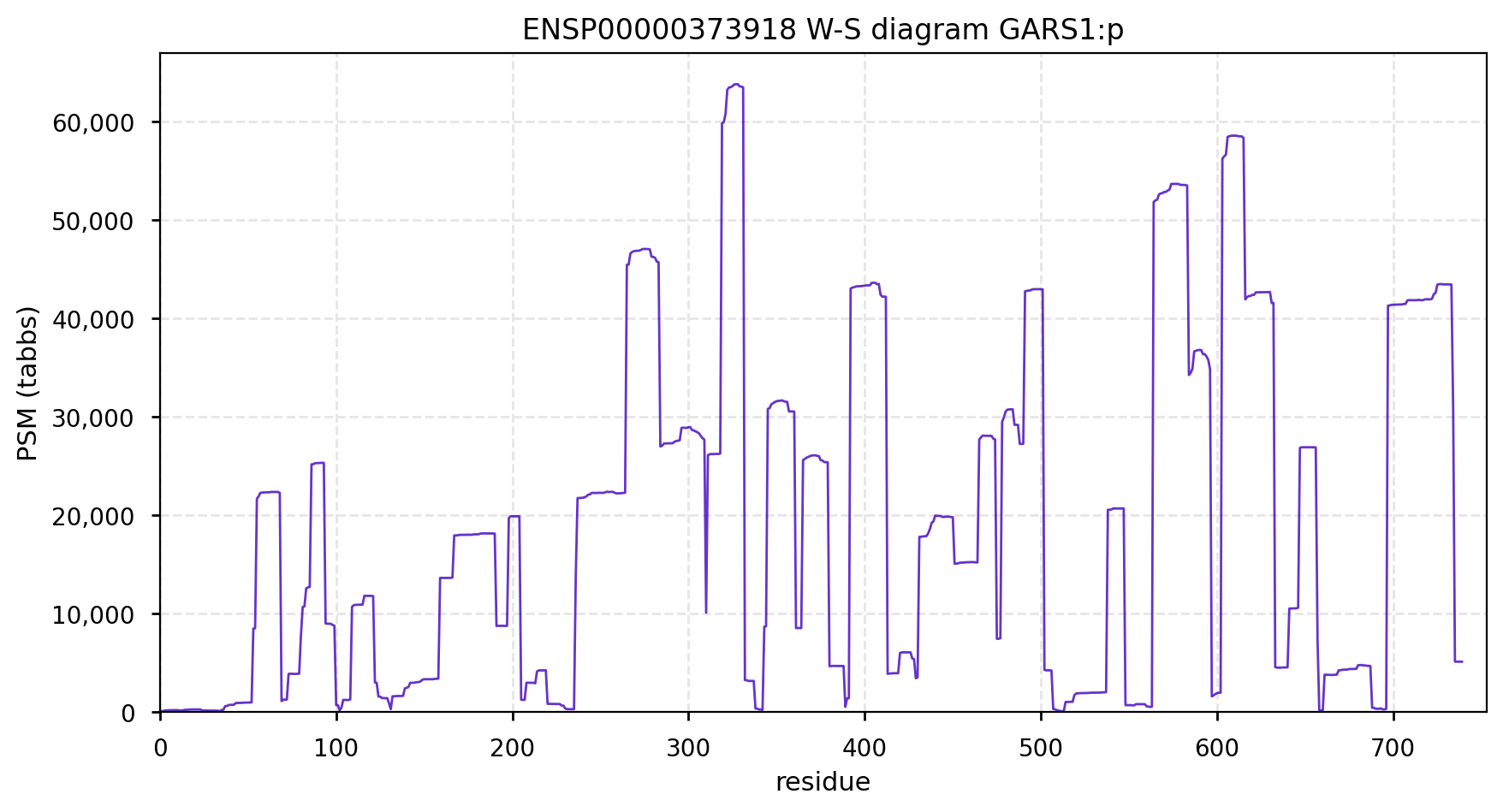 
Canon Folly: Palladin's ENSEMBL/MANE/INSDC/RefSeq canonical sequence (1223 aa) isn't detectable in vivo, while a shorter splice (777 aa) is widely distributed. In the images below, the observed coverage diagrams show the same 20 LC/MS/MS runs, fit to either the canonical or (777 aa) sequence. The 2 on top are bait-prey protein-protein interaction experiments with a recombinant canonical sequence as the bait. Human palladin, cytoskeletal associated protein (PALLD:p)  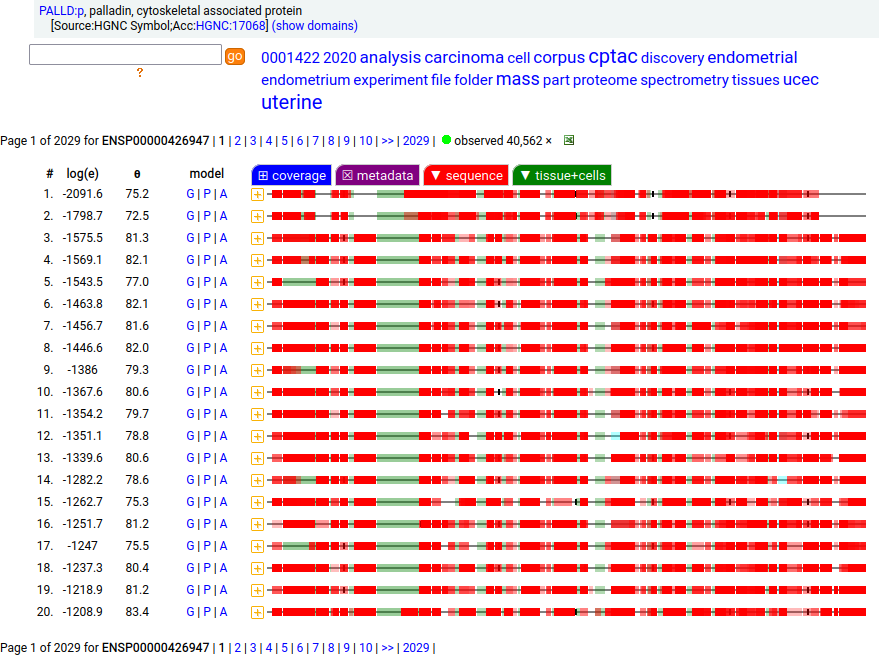
There is another splice variant (1106 aa) that appears to only be expressed in striated muscle (skeletal & cardiac). The (777 aa) form is used in smooth muscle (e.g., blood vessels, urinary bladder, colon). 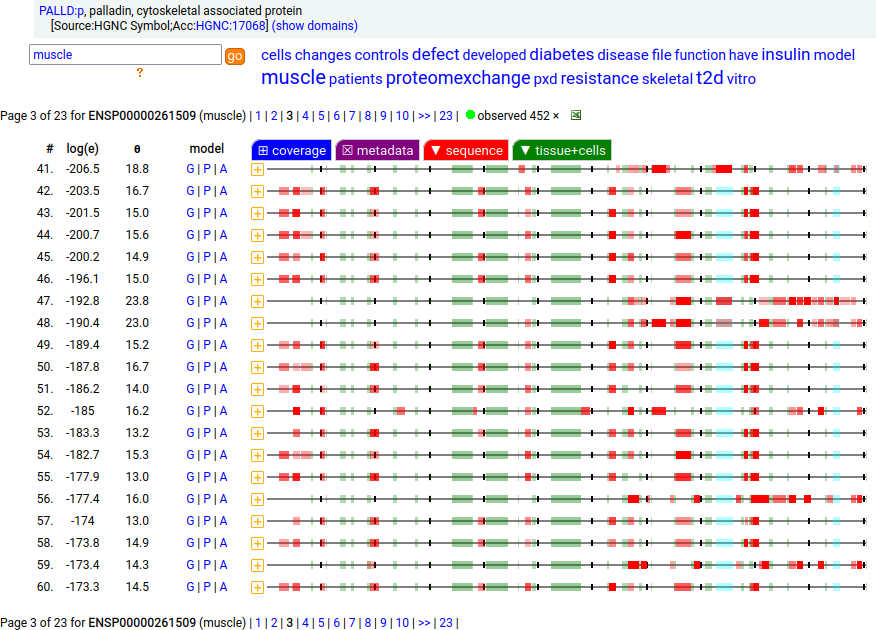
Canon Folly: FAM98B:p has a UP/ENSEMBL/MANE/CCDS/REFSEQ canonical sequence (433 aa) that is observed initiating at M1 (not acetylated), but a shorter version (422 aa) that initiates at M12 is acetylated and observed just as often in the same samples. The 3 peptides shown are useful to determine which initiation variants are present. It appears mammals all use this particular trick. Human family with sequence similarity 98, member B (FAM98B:p) 
Canon Folly: UniProt/ENSEMBL/MANE/CCDS/INSDC all agree that PNPO:g should result in the same canonical protein sequence (261 aa). Unfortunately the predominant observed protein (230 aa) is the result of translation initiation & N-acetylation at M32, the 2nd AUG in-frame on the mRNA. Human pyridoxamine 5'-phosphate oxidase (PNPO:p) 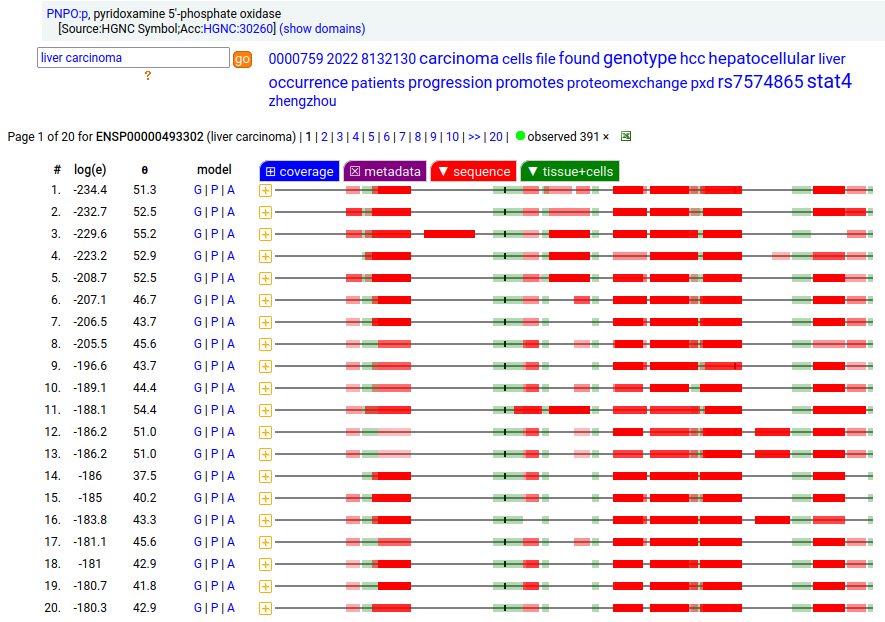
Canon Folly: Disagreements between canons is not unusual. REPIN1:p has two proposed canonical forms: ENSEMBL's with 2 exons (624 aa) and UNIPROT's 1 exon form (567 aa). The data has no observations of the peptides in the 1st exon of the 624 aa form , suggesting very strongly that the dominant translated "proteoform" is the 1 exon sequence. Human replication initiator 1 (REPIN1:p) 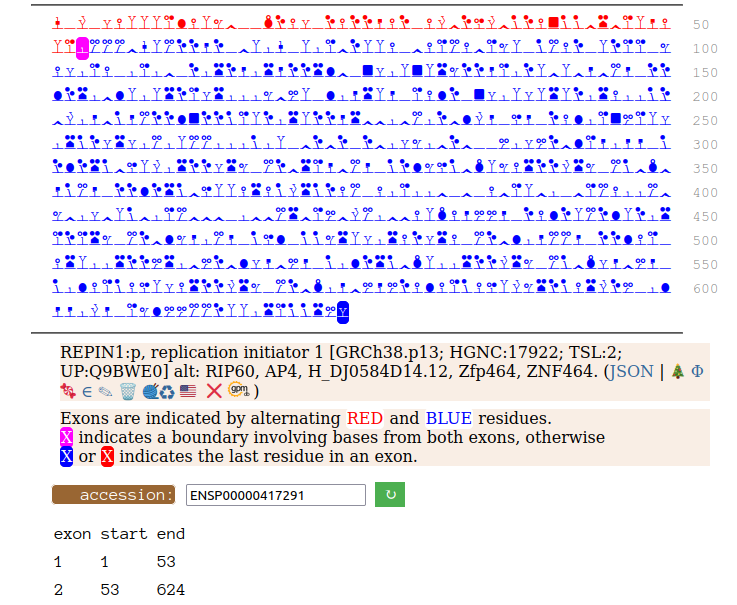 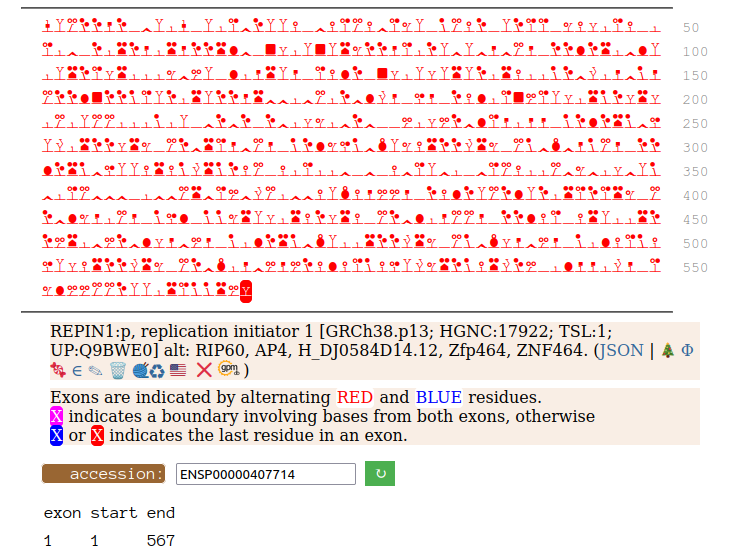
Canon Folly: NDUFV3:p is part of mitochondrial complex I. Its UP canonical sequence (108 aa) may be observed, but a different splice with an additional exon (473 aa) is much more common. Shown below are the coverage patterns observed for larger splice in BioPlex Interactome project data, with & without the UP canonical sequence as a bait. Human NADH:ubiquinone oxidoreductase subunit V3 (NDUFV3:p) 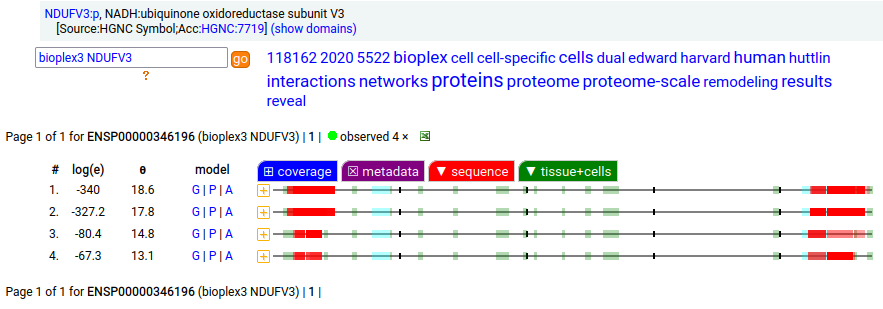 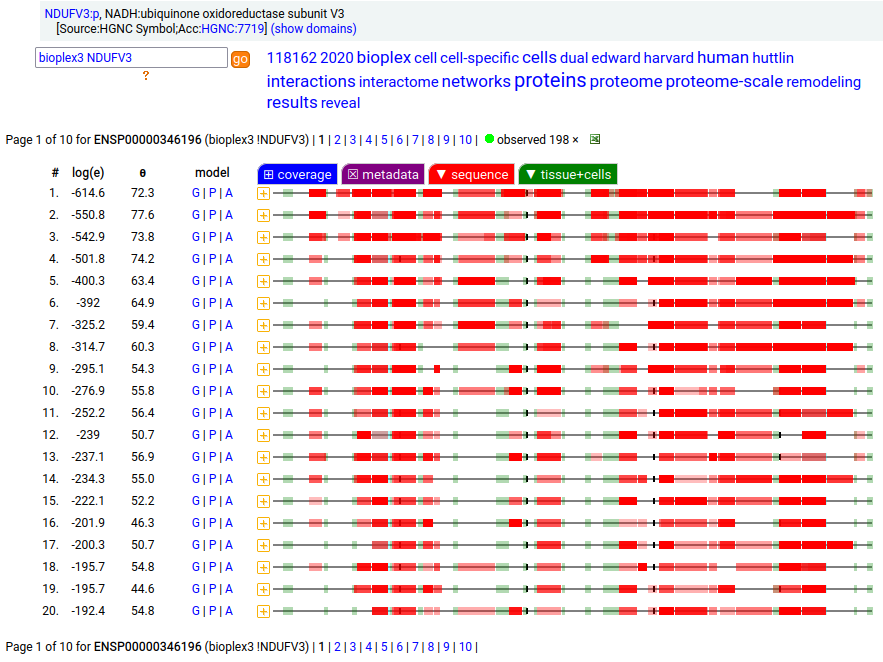
On the off chance that someone may be interested, here are the sequences of the 2 splices, with the exons marked.  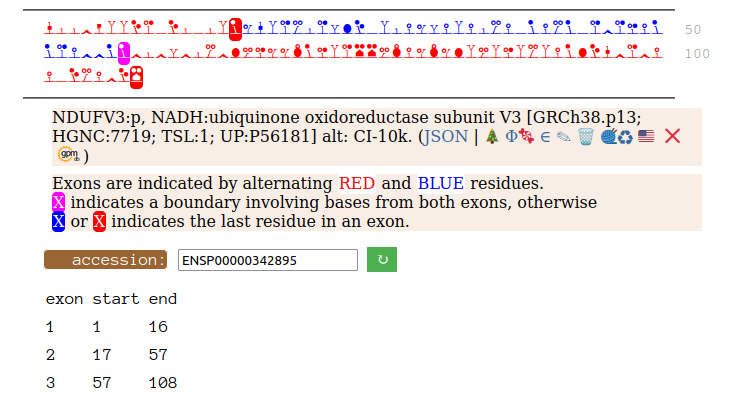
Canon Folly: ENSEMBL & UP canonical TSC22D1:p (1073 aa) is observed, but so is a much smaller splice (144 aa), which initiates at its M11. Both splices share 2 C-terminal exons, swapping out a single, long N-terminal exon (970 aa) for a short one (42 aa). BioPlex3 project data shows these 2 variants have different protein-protein interaction partners. Human transcription factor transforming growth factor β1-stimulated clone 22 domain family member 1 (TSC22D1:p) Canon Folly: The "canonical" sequence of UBAP2:p (1119 aa) is rarely translated, in favour of a subtly different, slightly shorter version (1117 aa). M1: canonical initiation site (1:20), co-translationally acetylated when this occurs Human ubiquitin associated protein 2 (UBAP2:p) Canon Folly: The "canonical" sequence (510 aa) of SYT6:p simply doesn't seem to exist in nature. Another GENCODE splice variant (425 aa) is closer, but the dominant sequence in tissue initiates at M30 rather than M1 of this splice (395 aa). The 425 aa sequence is seen occasionally as the bait in protein-protein interaction studies. Human synaptotagmin VI (SYT6:p)―GPMDB peptide identification diagram. 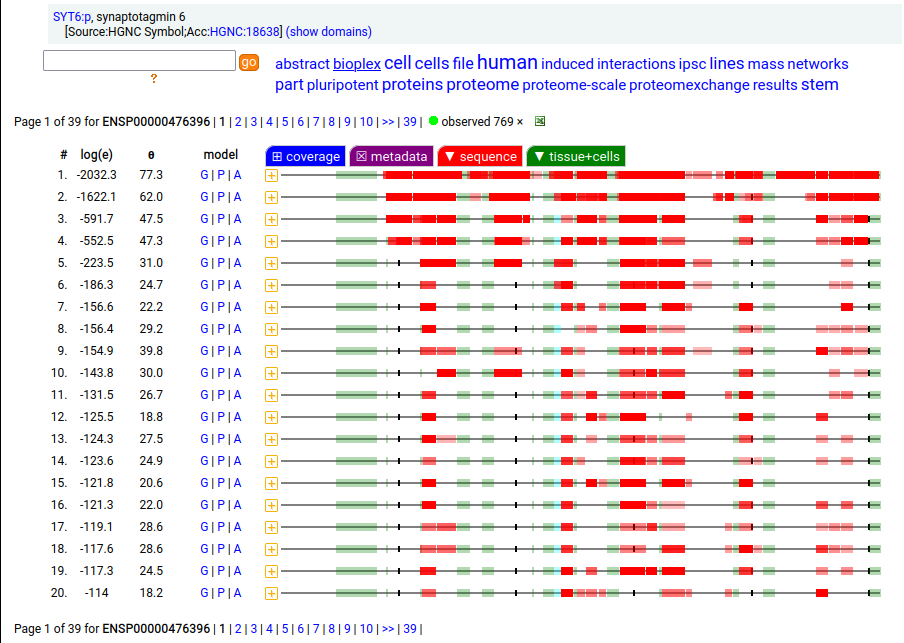 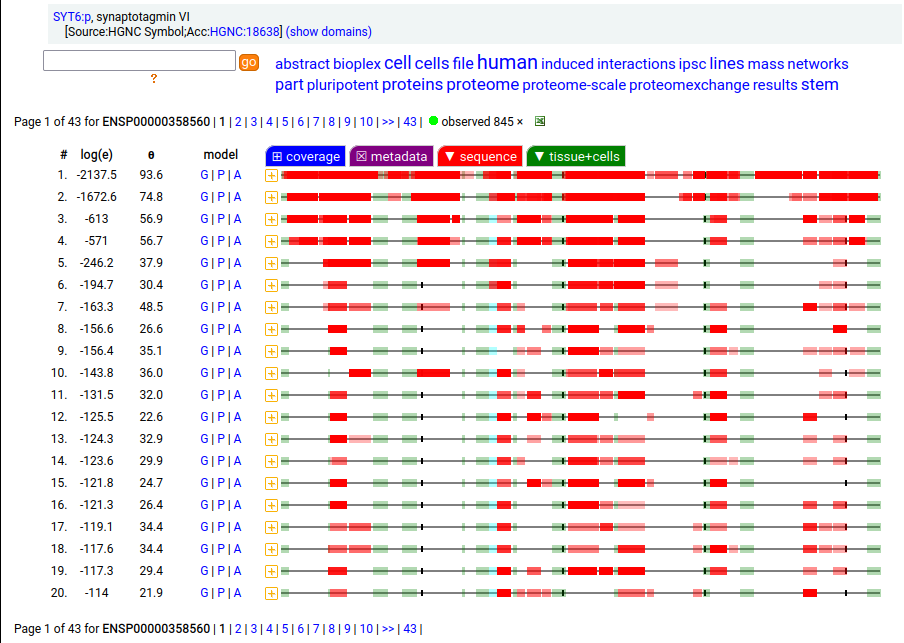
Canon Folly: which splice is the "right" splice? For the young, LEPR:p was one of the hottest targets in drug development for quite a while, but was also the ruin of many a scientist―it's ligand may have been the last nail in the coffin of the "protein engineering" craze back in the late 80's, early 90's.  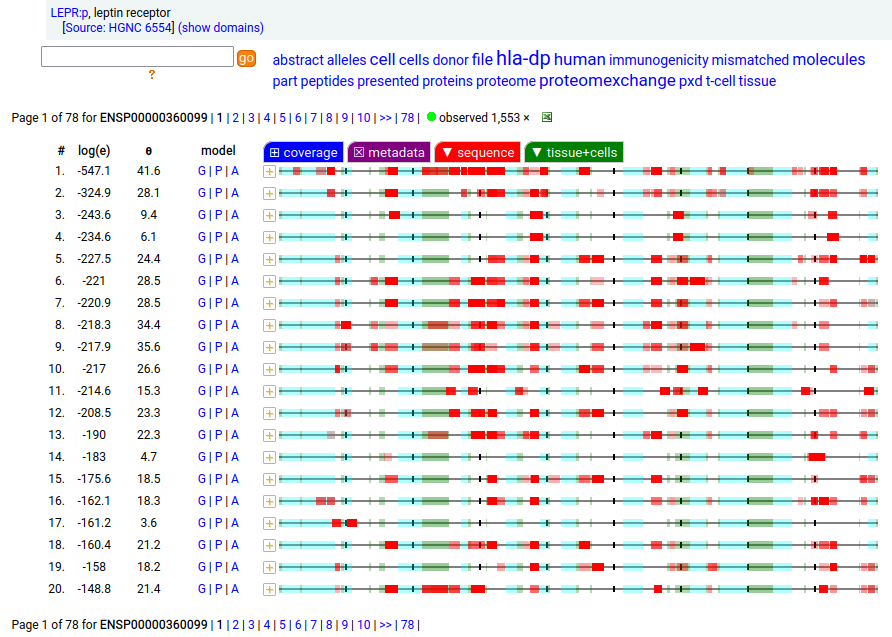
Proteins in the "cluster of differentiation" family are frequently found on
the surface of blood cells, where they are used for immunophenotyping (hence the name).
Many of them are type I membrane proteins, consisting of an extracellular N-terminal domain,
a transmembrane domain and an intracellular domain. While this is not the sort of
protein you might expect to see in urine, there are actually quite a few that are quite prominent in
urine MS/MS data. These are their stories.
A lymphocyte antigen that is a type I membrane protein with 1 extracellular immunoglobulin-like fold. Signal peptide: (1-30); extracellular region: (31-152); TM domain (153-175); intracellular SY-phospho-IDR (175-289). The soluble urine/blood plasma form does not have signals cward of K194. Human B and T lymphocyte associated (BTLA:p, :p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 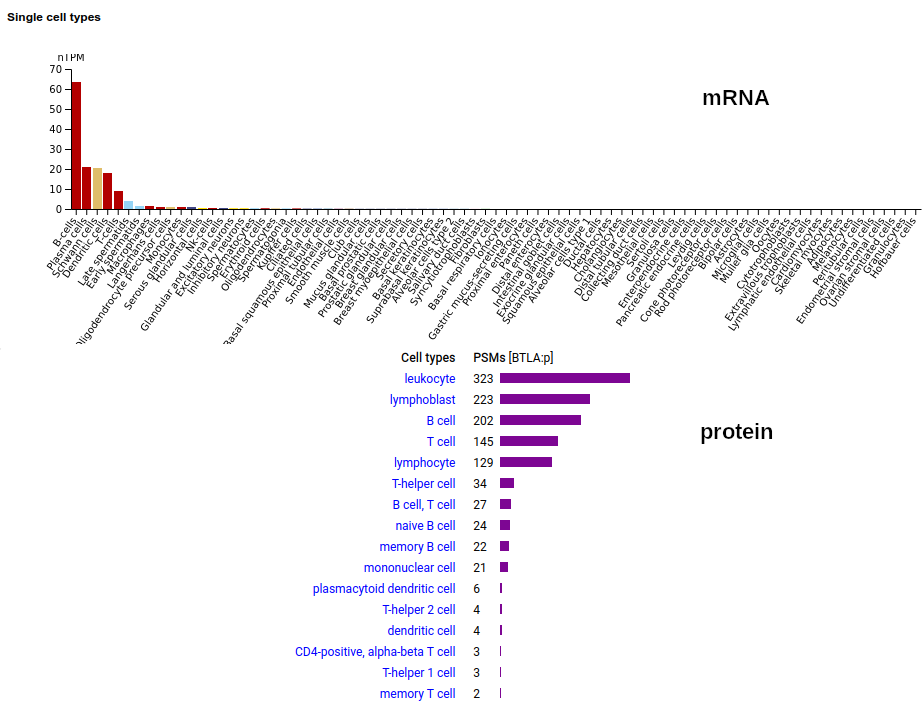 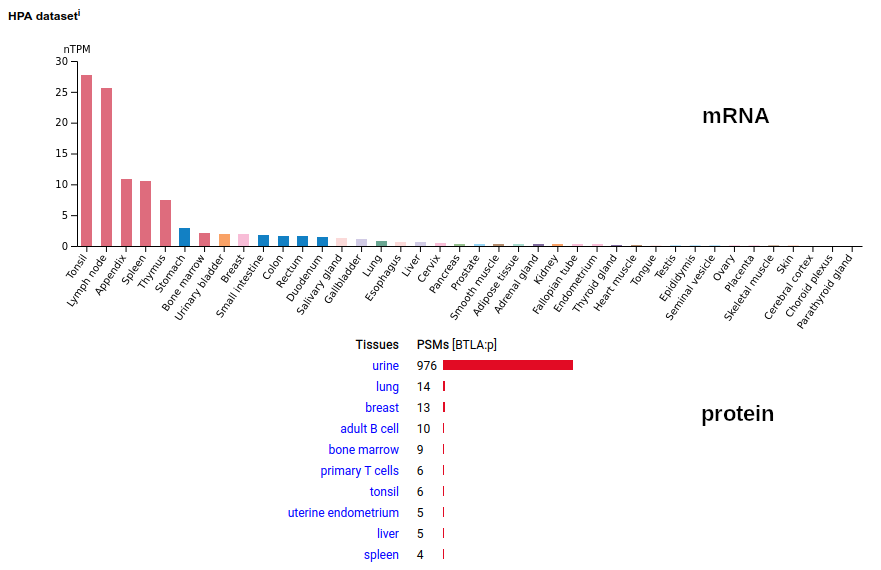 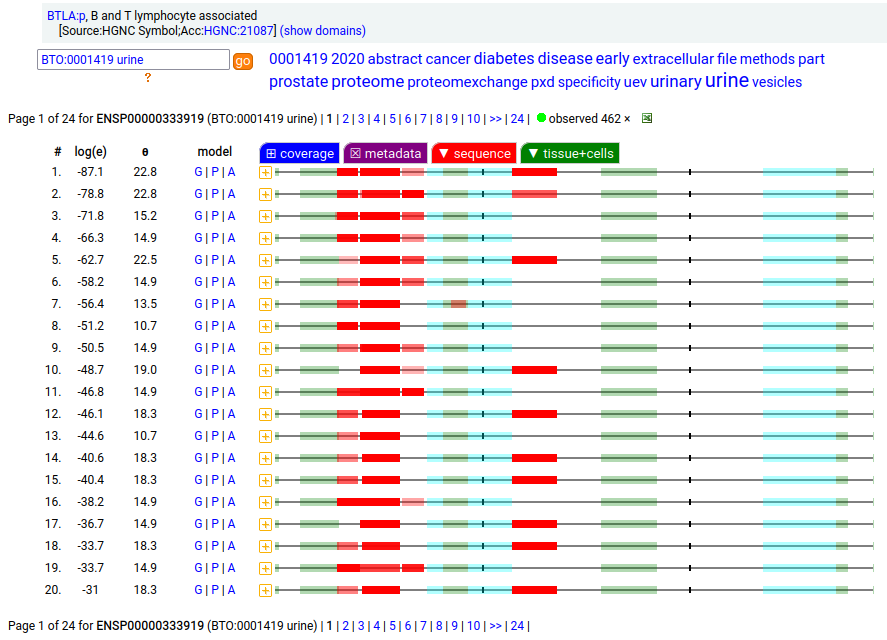 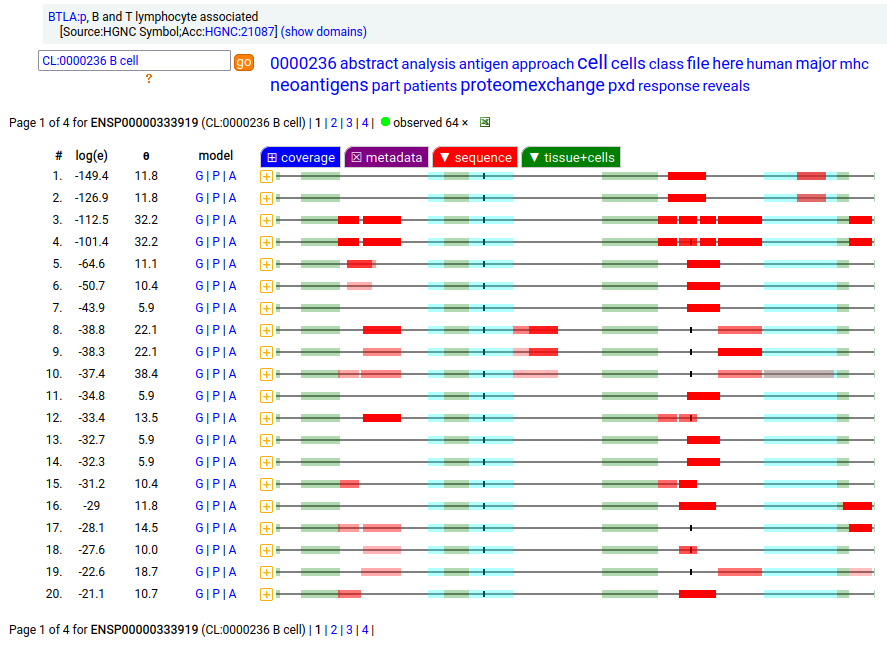
A tissue antigen that is a type I membrane protein with 1 extracellular TNF receptor domain. Signal peptide: (1-28); extracellular region: (29-249); TM domain (250-272); intracellular ST-phospho-IDR region (273-427). The soluble urine/amniotic fluid form does not have signals cward of R194. Human nerve growth factor receptor (NGFR:p, CD271:p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 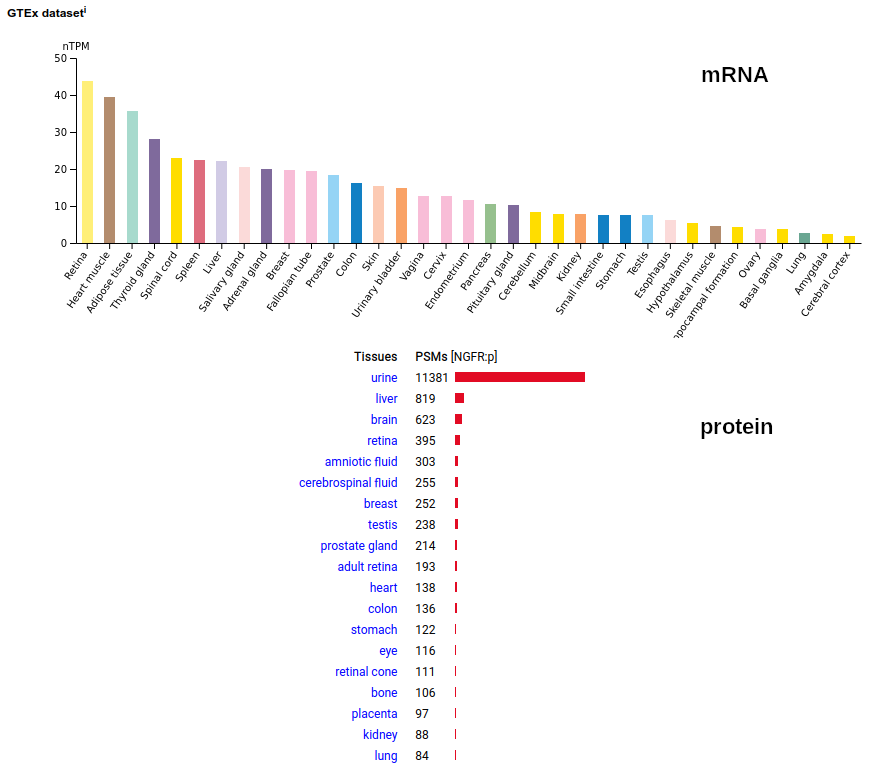 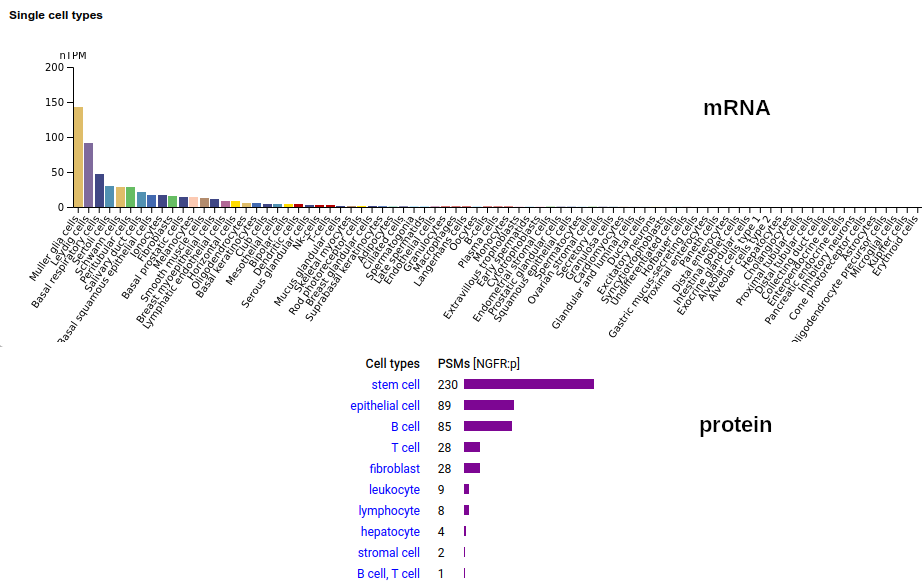  
This protein is a type I cell membrane protein, with a single TM domain (686-708) and a pyro-Glu N-terminus formed at Q18 following the removal of its signal peptide. This form dominates in solid tissues & cells. In fluids, the TM & intracellular regions have been removed creating an observable soluble form (C-terminus: L655). Human cluster of differentiation member 248 (CD248:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 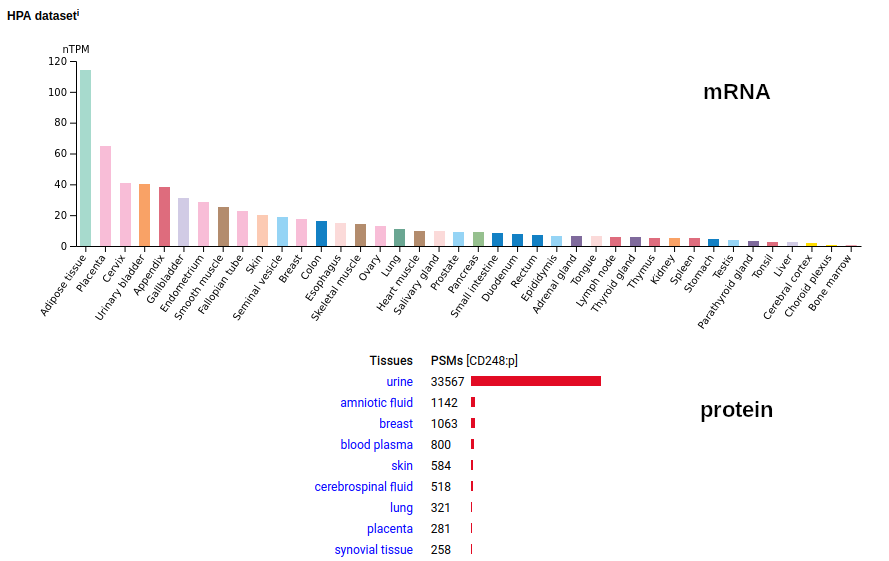
This soluble form is actually a little large to end up in the urine―it is a bit bigger than serum albumin―but it does end up being concentrated there pretty routinely so it must be able to squeak its way through the glomerular filtration apparatus somehow. This fibroblast antigen is a type I membrane protein with 1 extracellular F5/8 type C domain. Signal peptide: (1-23); extracellular region: (24-498); TM domain (399-421); intracellular SY-phospho-IDR+kinase region (422-855). The soluble urine/blood plasma form does not have signals cward of R341. Human discoidin domain receptor tyrosine kinase 2 (DDR2:p, :p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 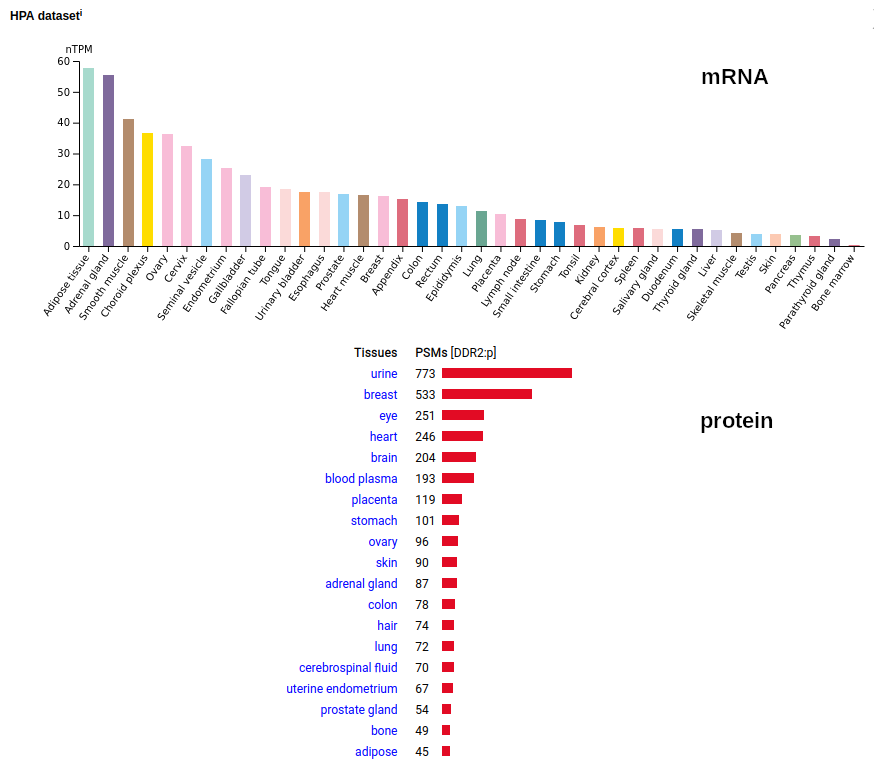 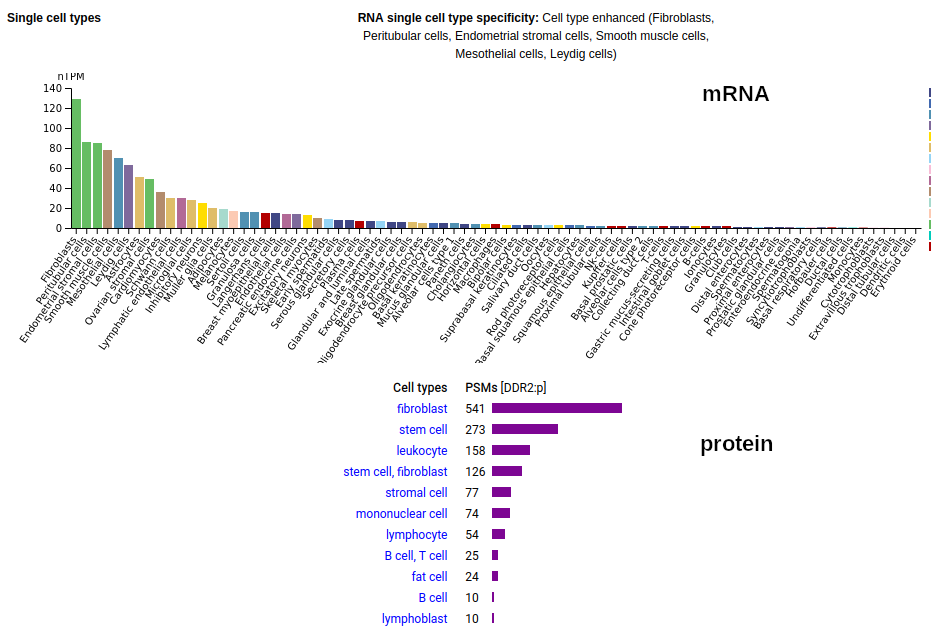 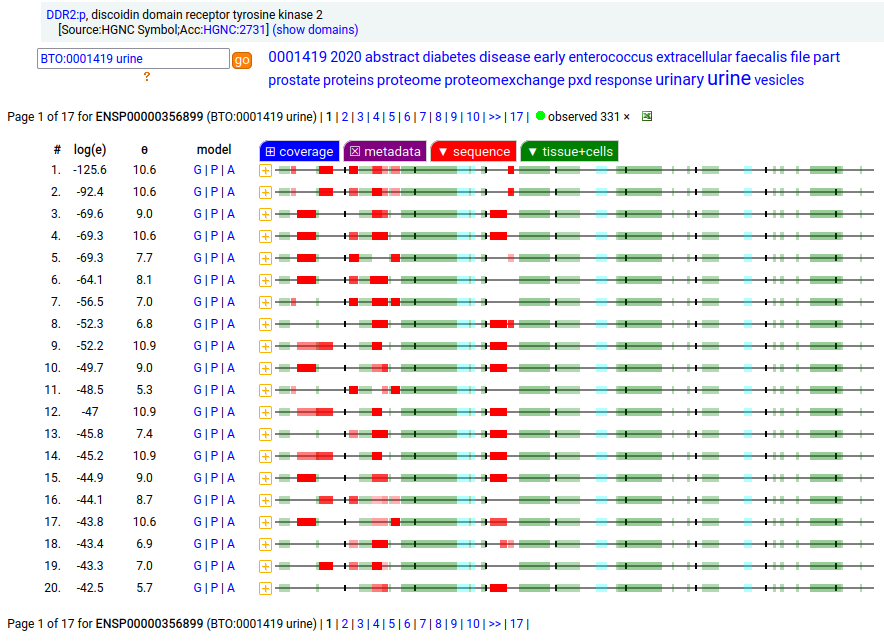 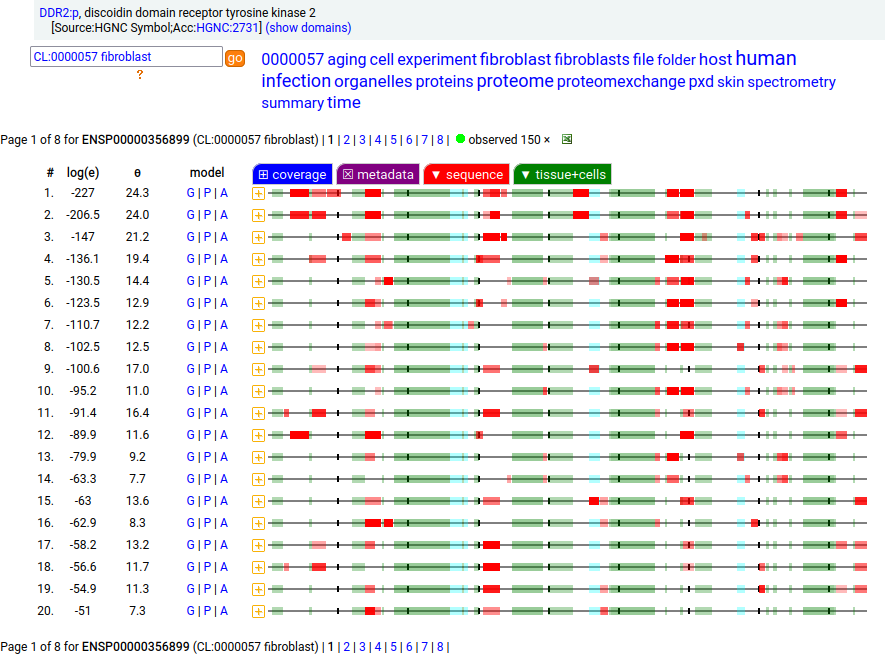
DDR2:p has an additional SP-type acceptor, presumably needed to deal with cell-cycle related issues that aren't a factor for DDR1:p: 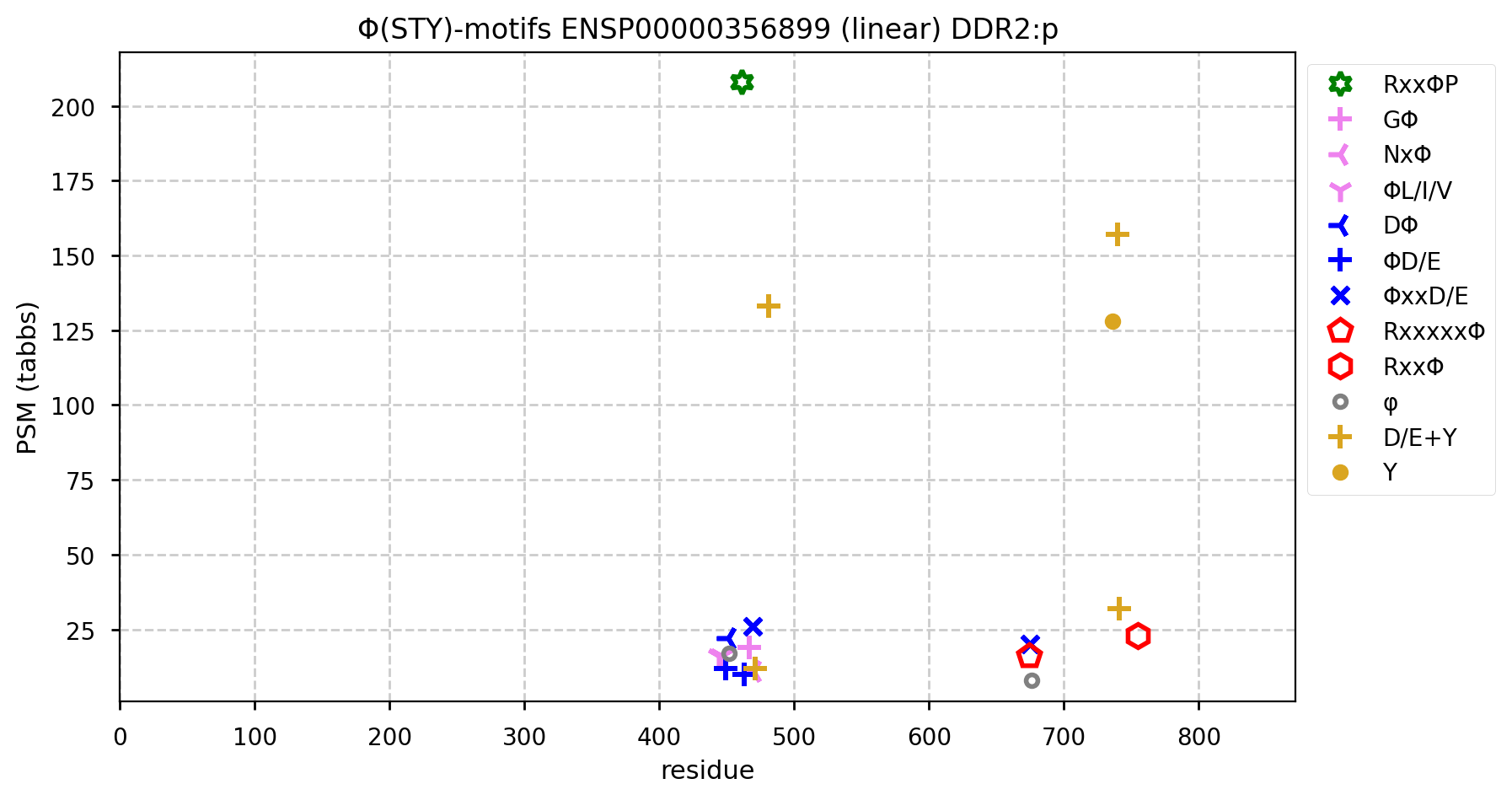
This antigen is a type I membrane protein with 1 extracellular F5/8 type C domain. Signal peptide: (1-20); glycosylated extracellular region: (21-416); TM (417-439); intracellular Y-phospho-IDRs + kinase domain (440-913). The soluble urine/milk/blood plasma form does not have signals cward of R341. Human discoidin domain receptor tyrosine kinase 1 (DDR1:p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 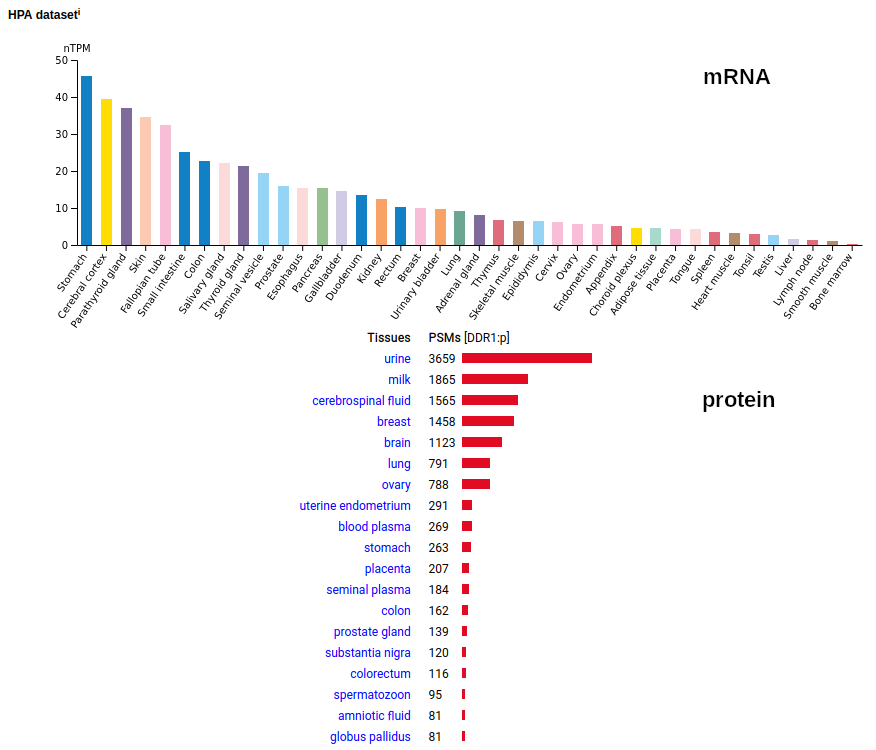 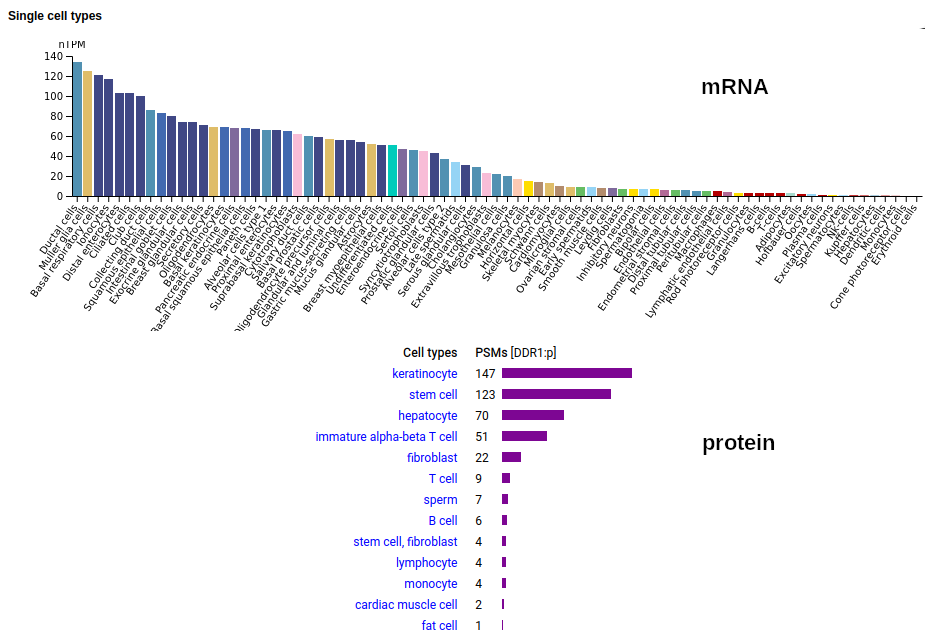 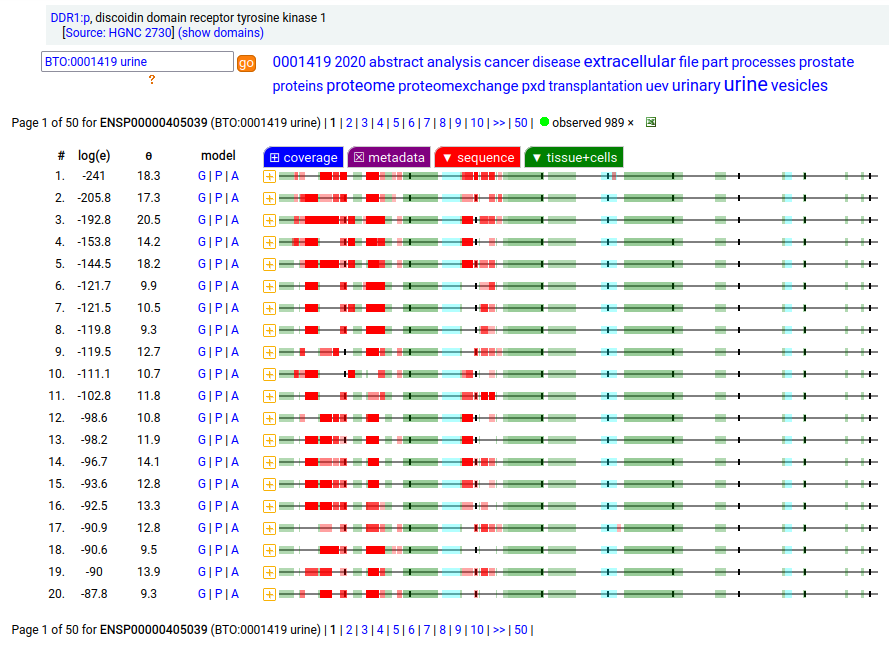 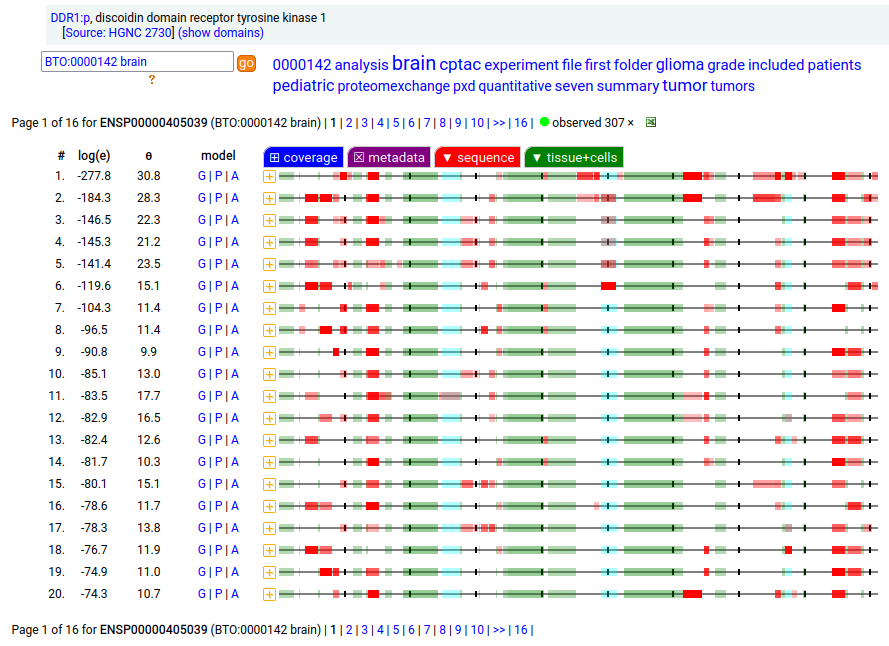
The Y+phosphoryl pattern associated with the intracellular region show multiple acceptors, some potentially auto-phosphorylated. 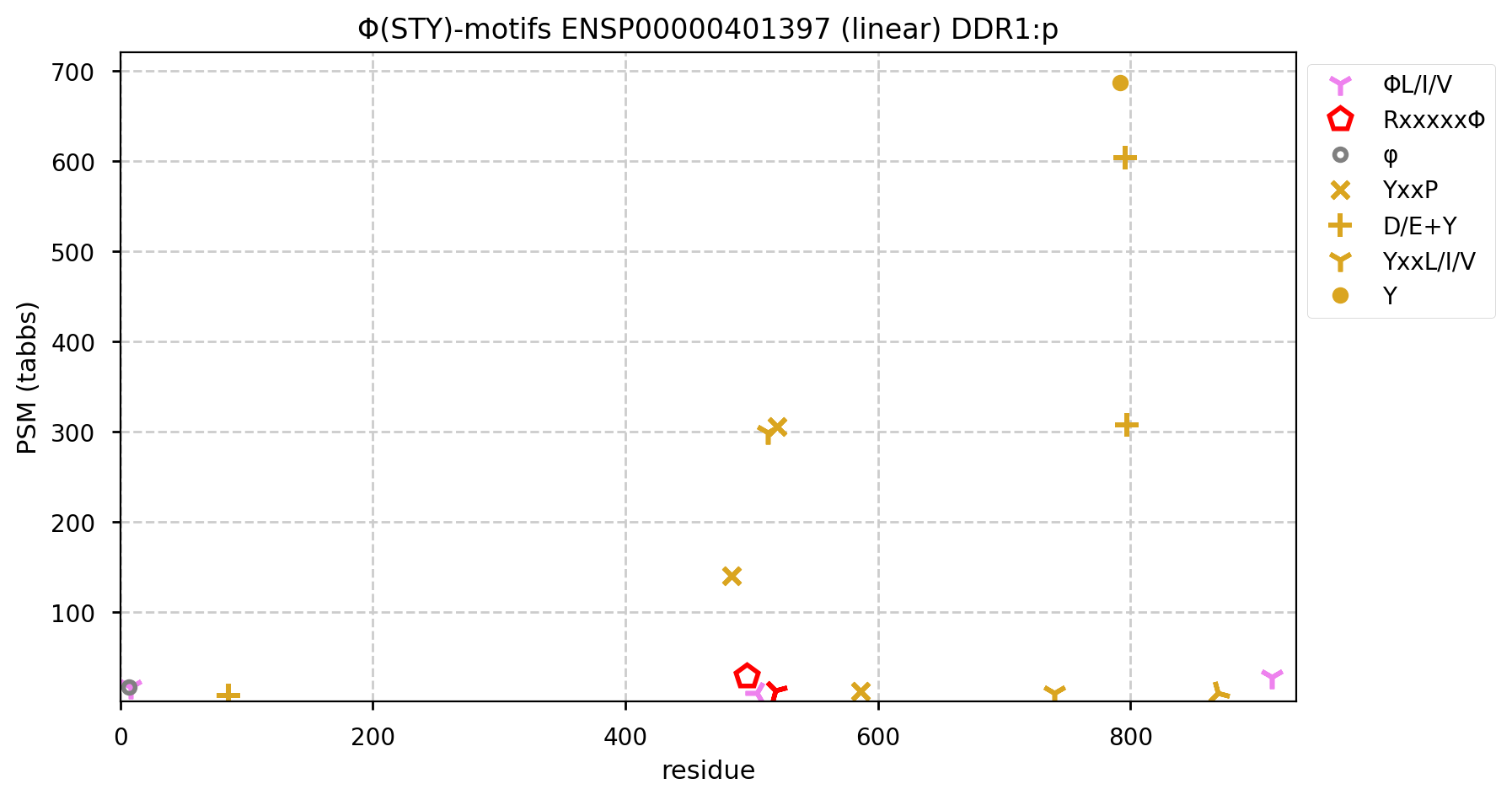
This lymphocyte antigen is a type I membrane protein with 2 extracellular immunoglobulin-like folds. Signal peptide: (1-20); glycosylated extracellular region: (21-237); TM domain (238-260); intracellular Y-phospho-IDR (261-335). The soluble urine form has no signals cward of K147. Human signaling lymphocytic activation molecule family member 1 (SLAMF1:p, CD150:p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 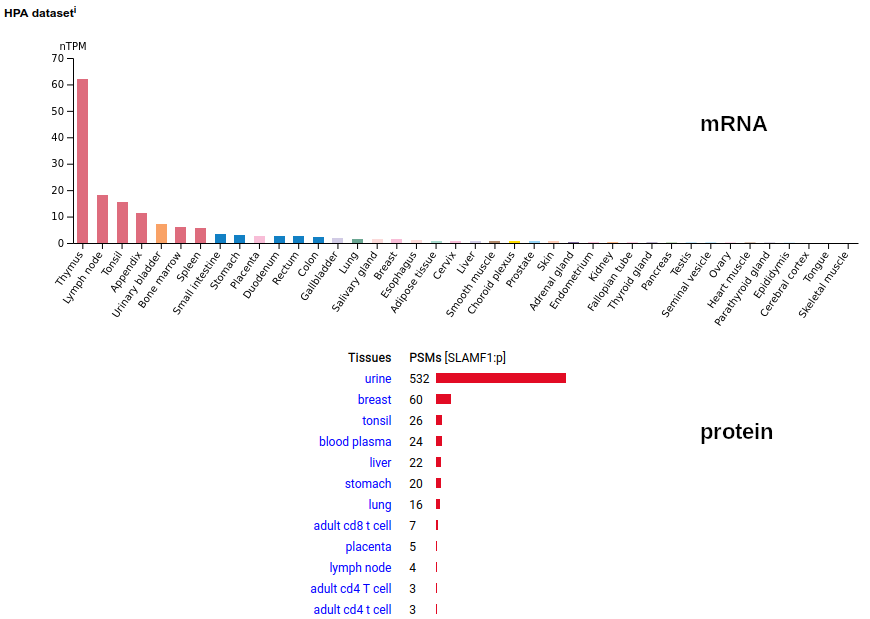 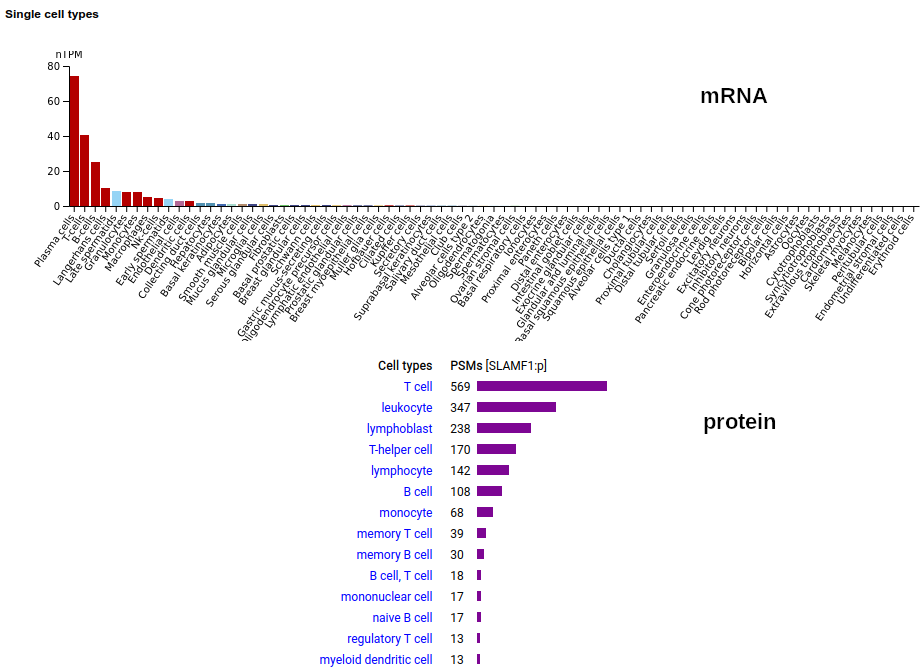 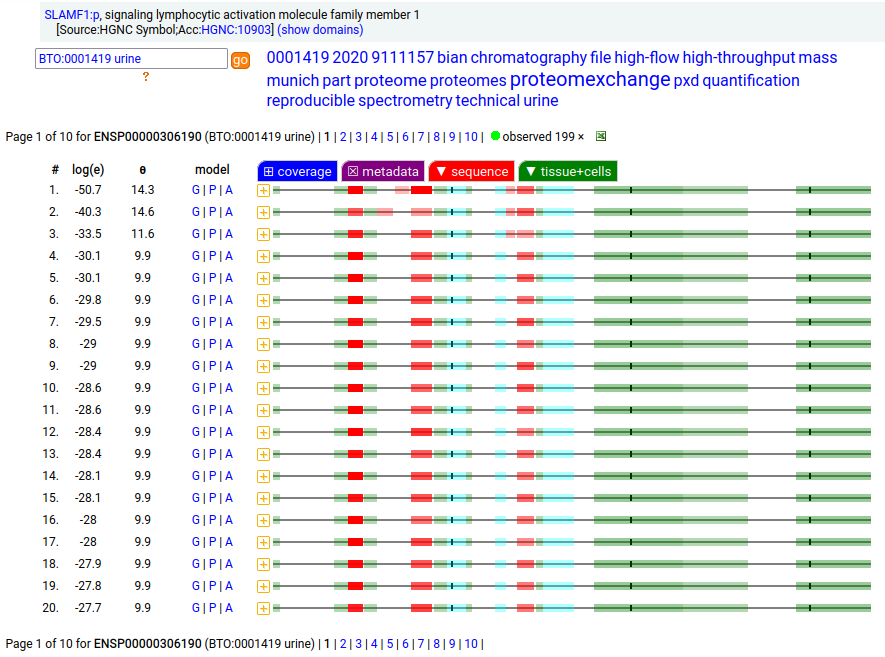 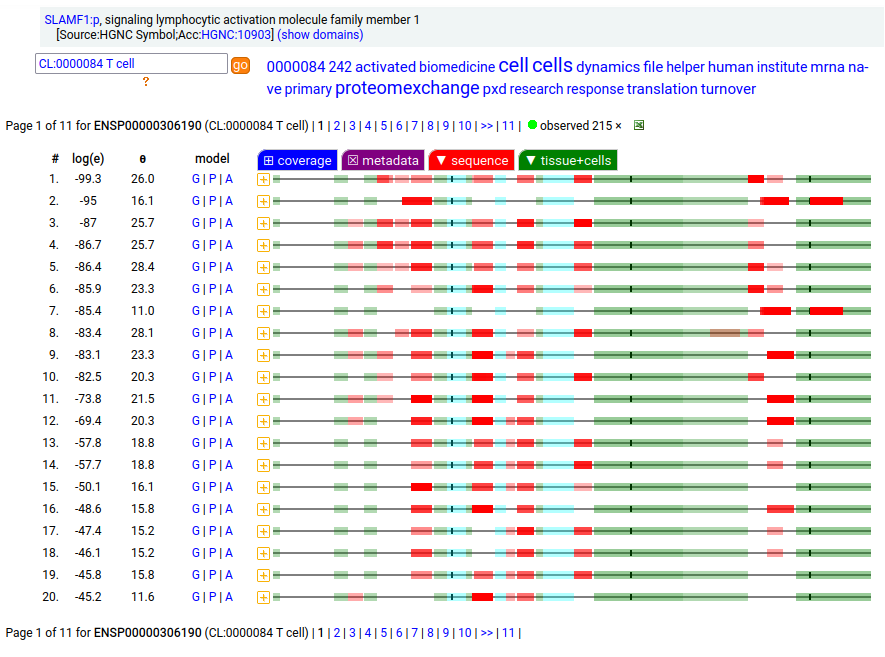
This solid tissue surface antigen is a type I membrane protein with 5 extracellular immunoglobulin-like folds. Signal peptide: (1-23); N-glycosylated extracellular region: (24-559); TM domain (560-582); intracellular SY-phospho-IDR (583-646). The soluble urine/blood/CSF form does not have signals cward of K507. Human melanoma cell adhesion molecule (MCAM:p, CD146:p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 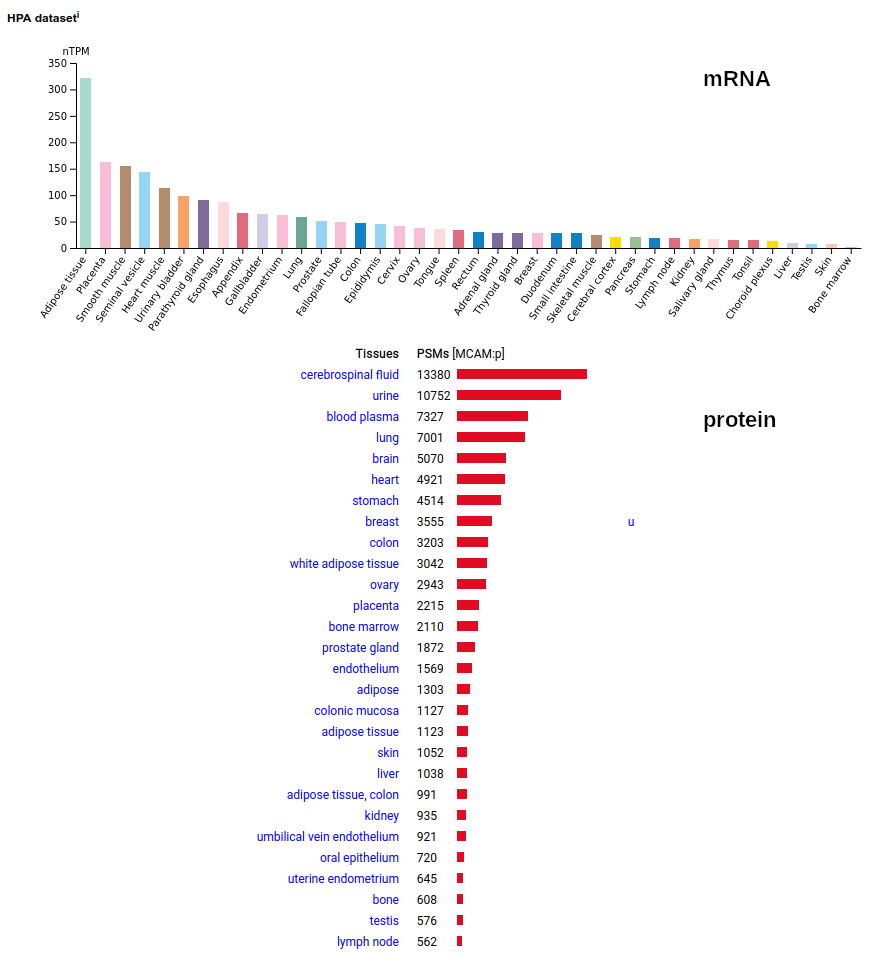 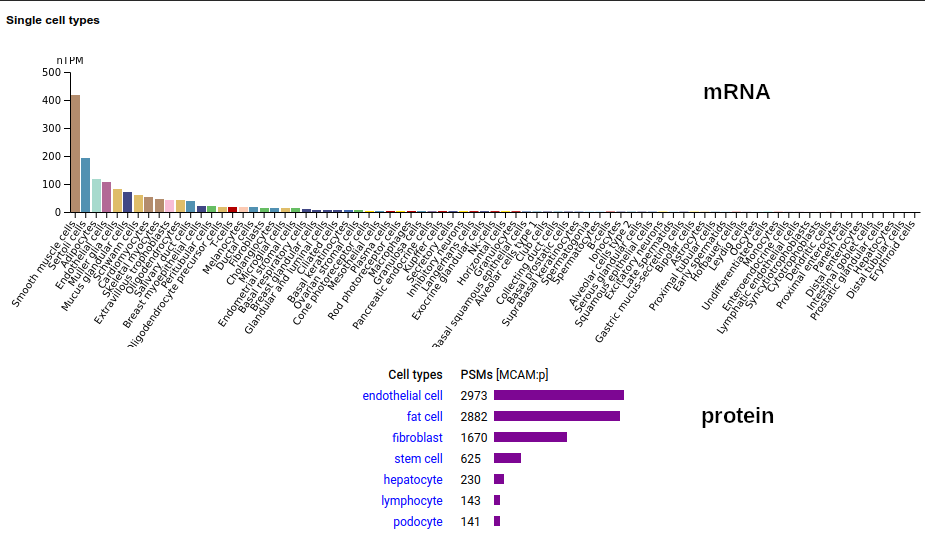 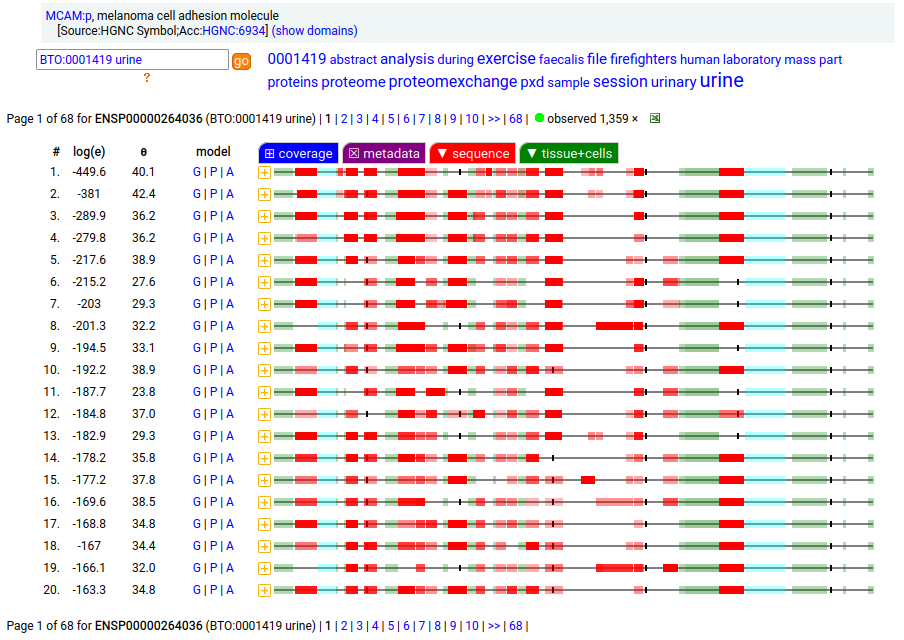 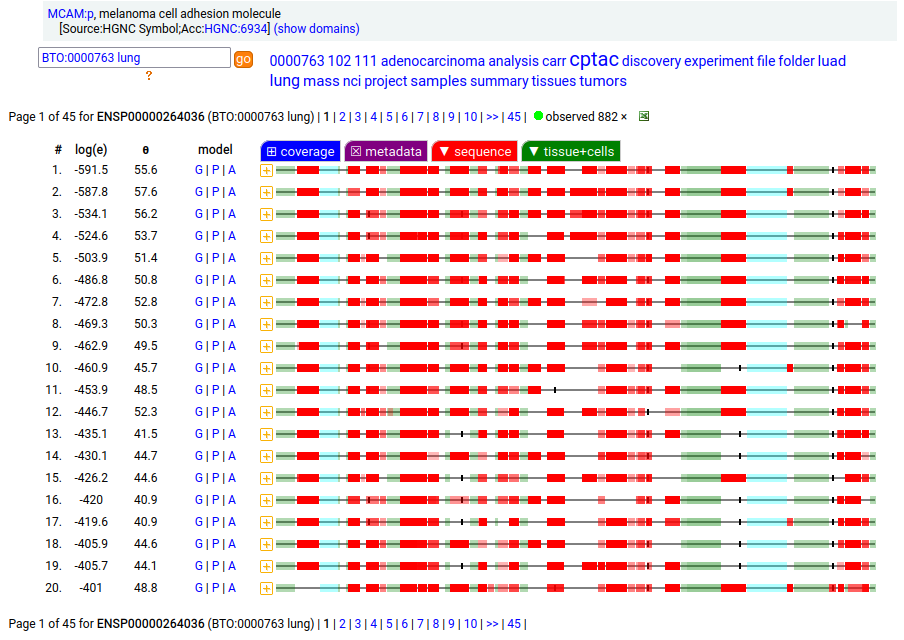
This T cell antigen is a type I membrane protein with 2 extracellular immunoglobulin-like folds. Signal peptide: (1-26); extracellular region: (27-242); TM domain (243-265); intracellular S-phospho-IDR (266-551). The soluble urine form only has rare signals cward of R230. Human interleukin 2 receptor subunit beta (IL2RB:p, CD122:p)―HPA mRNA tissue & cell type distributions paired with GPMDB protein tissue & cell type tabbulations. 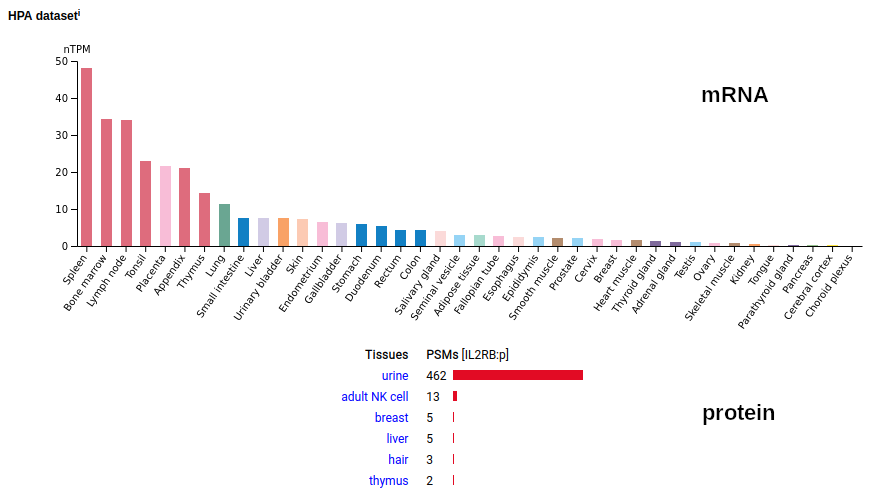 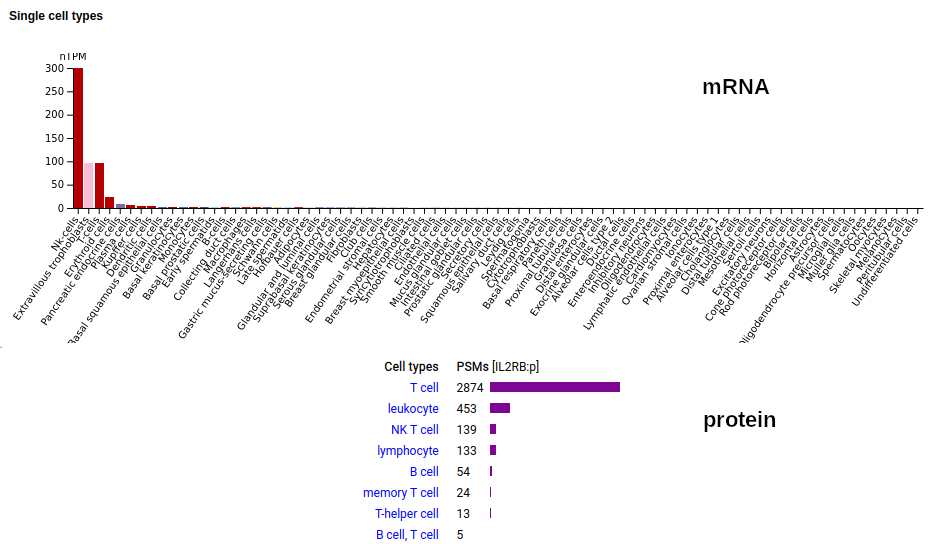
The peptide signal patterns generated by the soluble (urine) form is clearly different than the intact form found in T-cell samples. 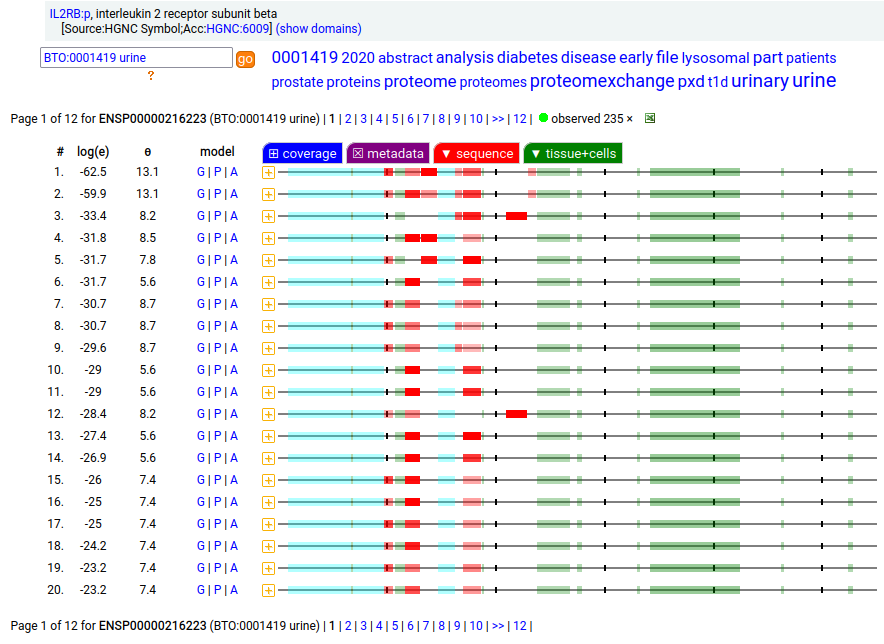 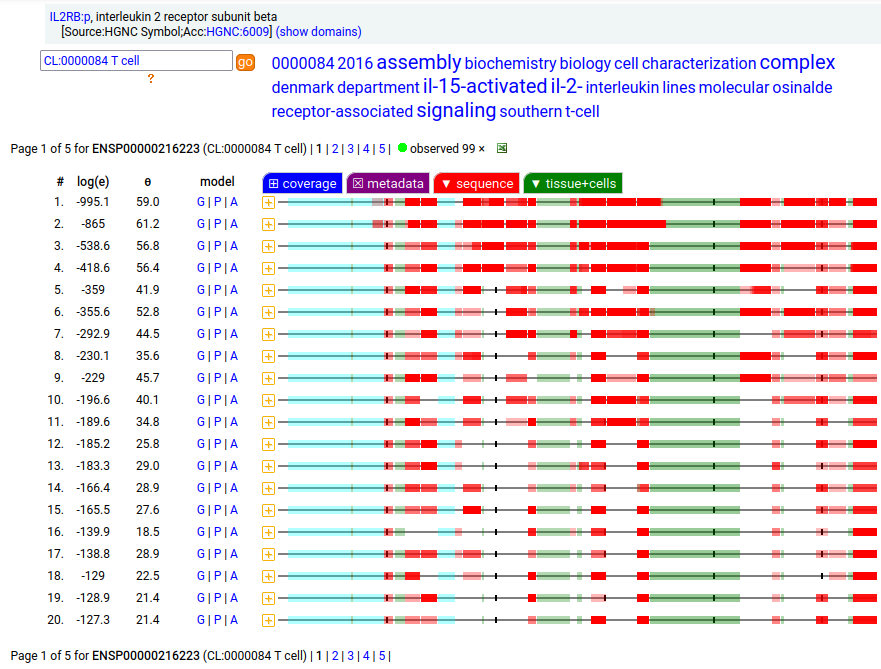
While CD99L1:g turned out to be a pseudogene, this protein is a type I membrane protein without extracellular folds. Signal peptide: (1-[24-28]); extracellular region: ([25-29]-185); TM domain (186-208); intracellular phospho-IDR (209-262). The soluble urine form only has rare signals cward of K163. Human cluster of differentiation member 99-like 2 (CD99L2:p)―HPA mRNA tissue & GPMDB protein tissue tabbulations. 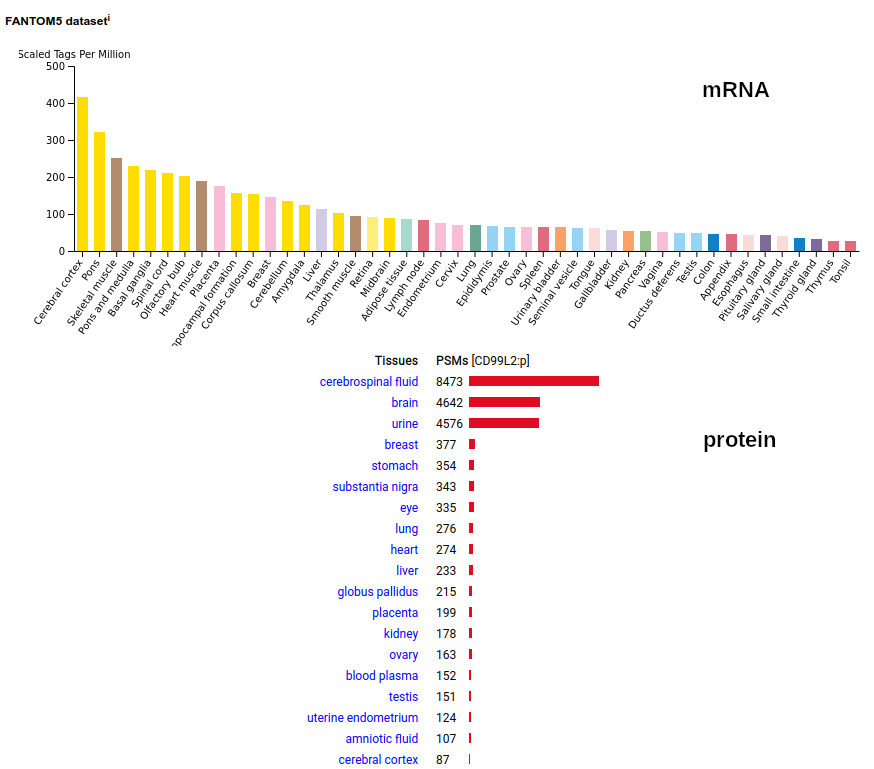
The intact, membrane embedded form of CD99L2:p is most abundant in CNS samples, including cerebrospinal fluid. In these samples, the intact TM domain may be observed as part of the tryptic peptide (170-211): r . YGSNDDPGSGMVAEPGTIAGVASALAMALIGAVSSYISYQQK . k The truncated, soluble urine form occasionally extends as far cward as A189, but never includes the TM-containing peptide. 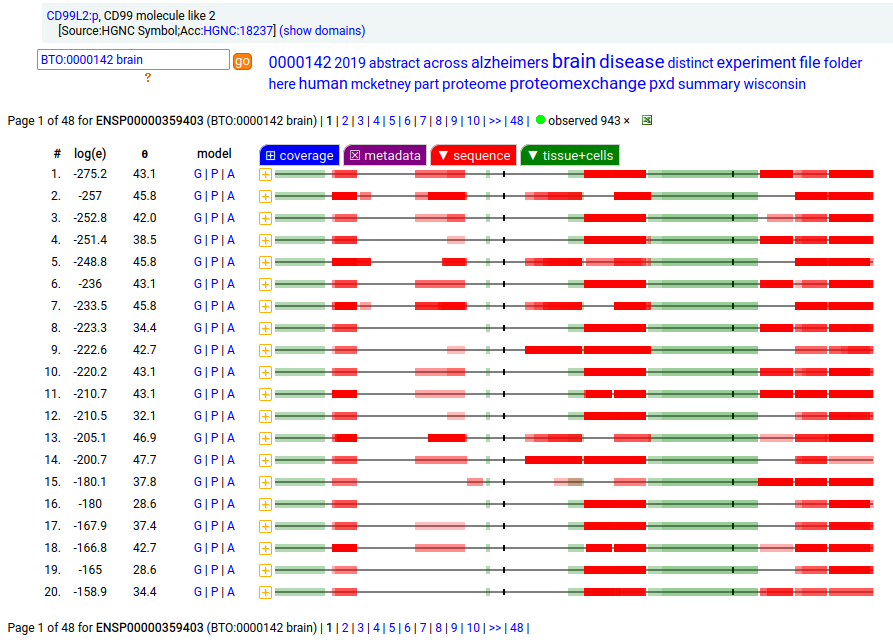 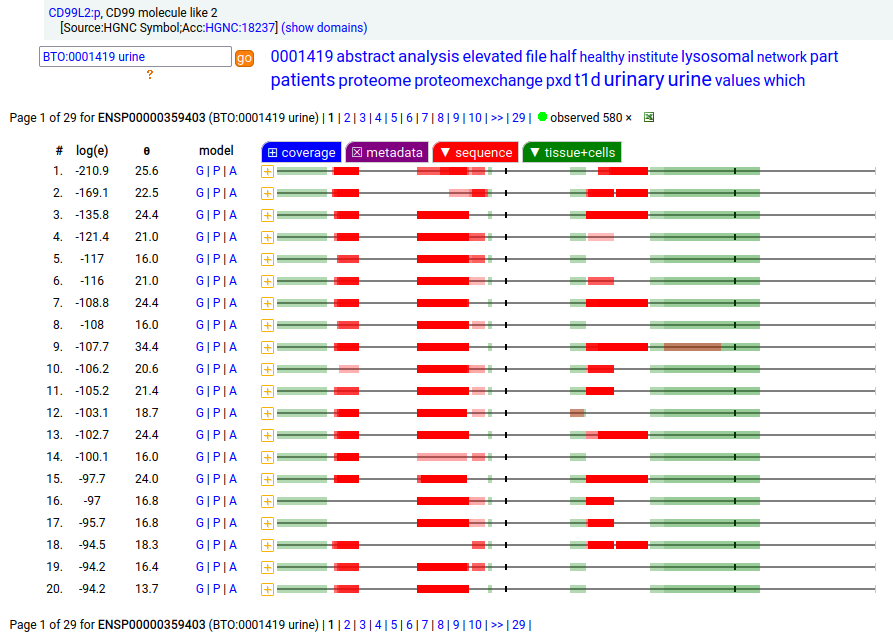
Part of the largely forgotten XG blood group of antigens, this one is a type I membrane protein without any extracellular structure. Signal peptide: (1-21); extracellular region: (22-125); TM domain (126-148); intracellular ST-phospho-IDR (149-185); T41, S48+glycosyl. The soluble form in urine has no signals cward of V135. Human cluster of differentiation member 99 (CD99:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations.  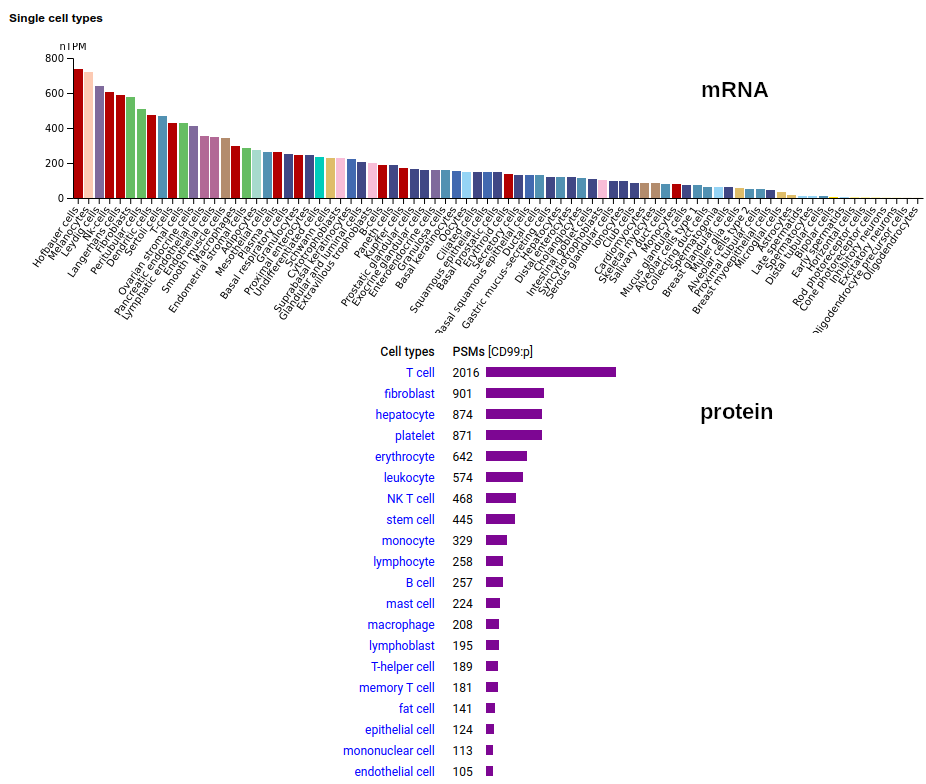
The coverage diagrams for "urine" vs "HeLa" demonstrates a soluble form that is truncated at the TM domain. The TM domain here is part of an observable tryptic peptide (117-150) that is eluted just before the gradient ends in many common LC methods: k . EGEEADAPGVIPGIVGAVVVAVAGAISSFIAYQK . k 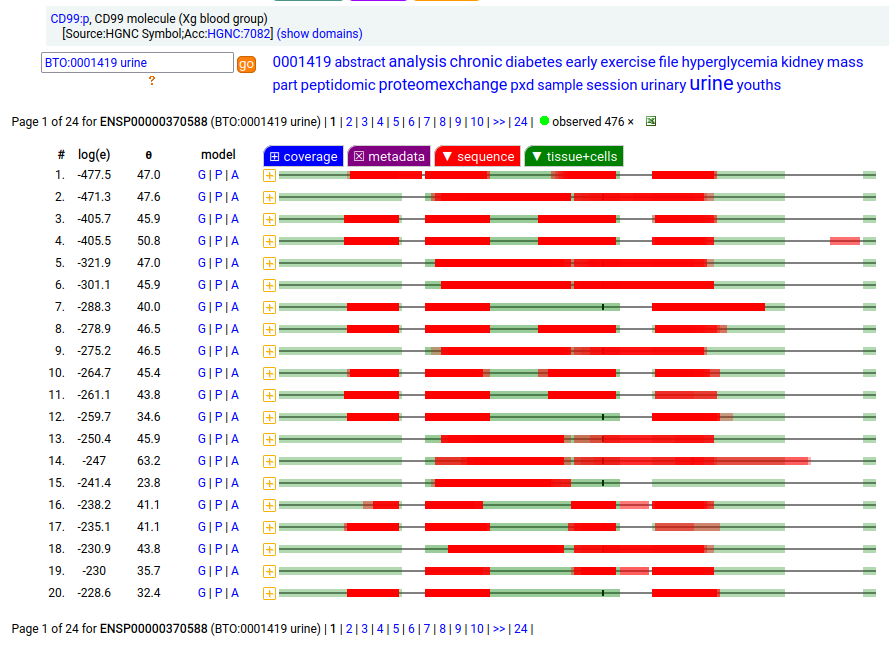 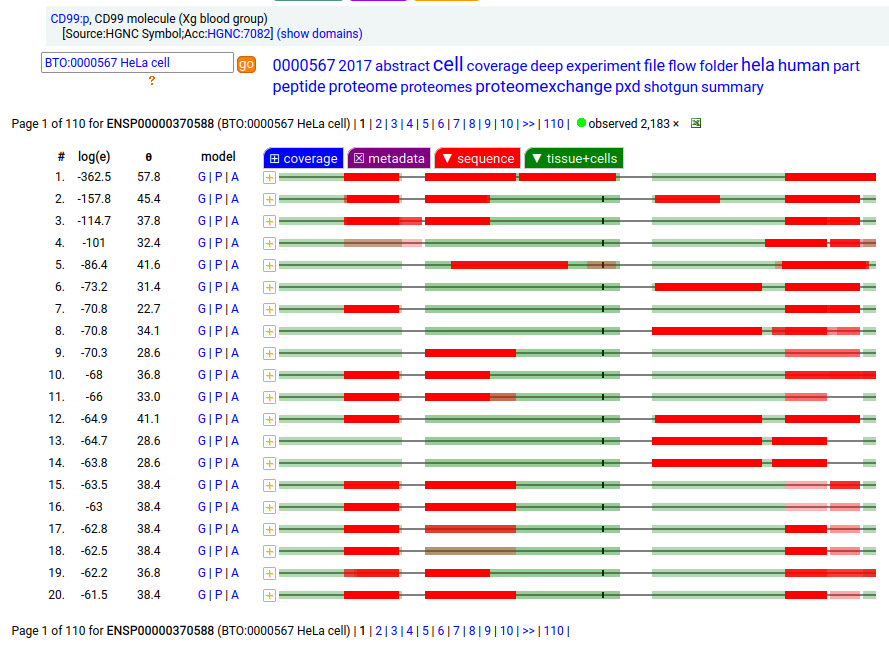
FCAR:p may be found in the monocyte lineage: a type I membrane protein with 2 extracellular Immunoglobulin-like folds. Signal peptide: (1-21); extracellular region: (22-227); TM domain (228-246); intracellular S-phospho-IDR (247-287); N79, N141, N186+glycosyl. The soluble form in urine has no signals cward of R230. Human cluster of differentiation member 89 (CD89:p/#FCAR:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations. 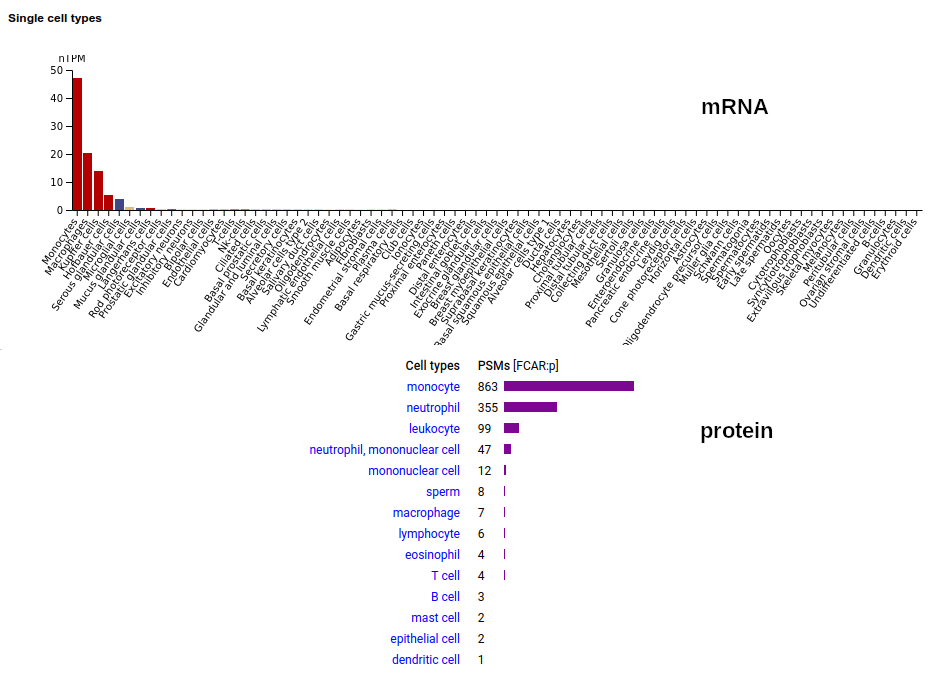 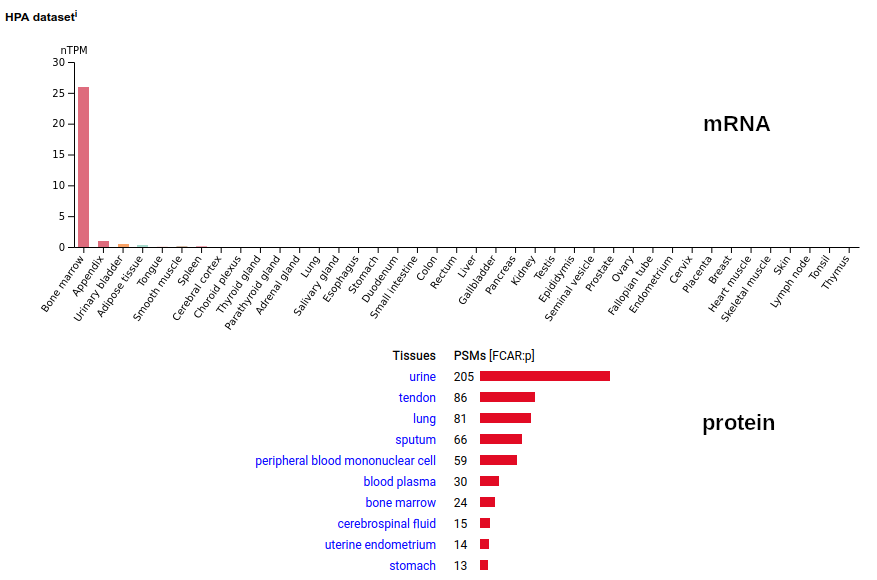
The coverage diagrams for "urine" vs "monocyte" demonstrate the loss of the TM & intracellular regions in the soluble form. In order for the membrane form to function (binding IgA), it must bind to one of the Fc-gamma receptors, e.g. CD32 or CD64. 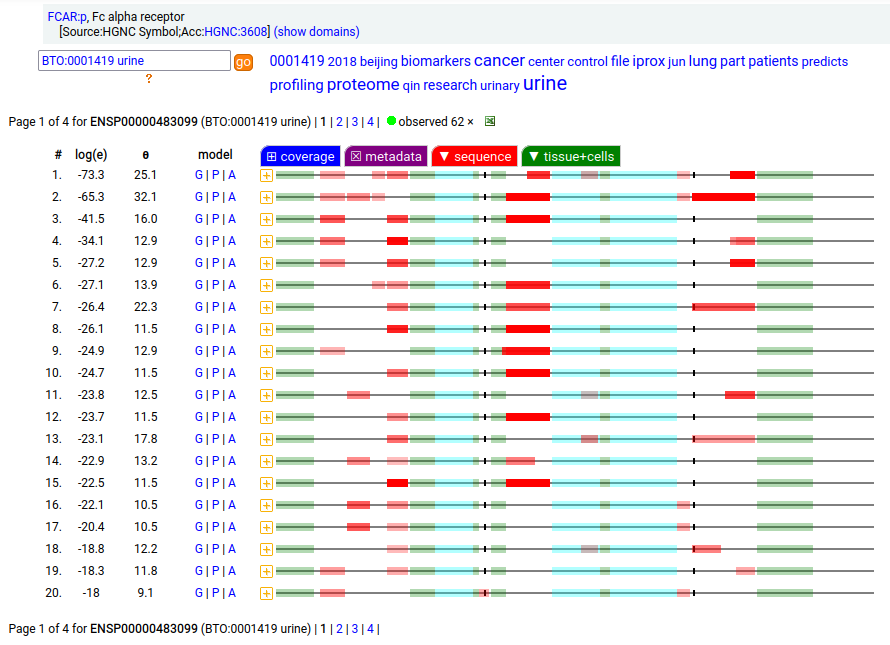 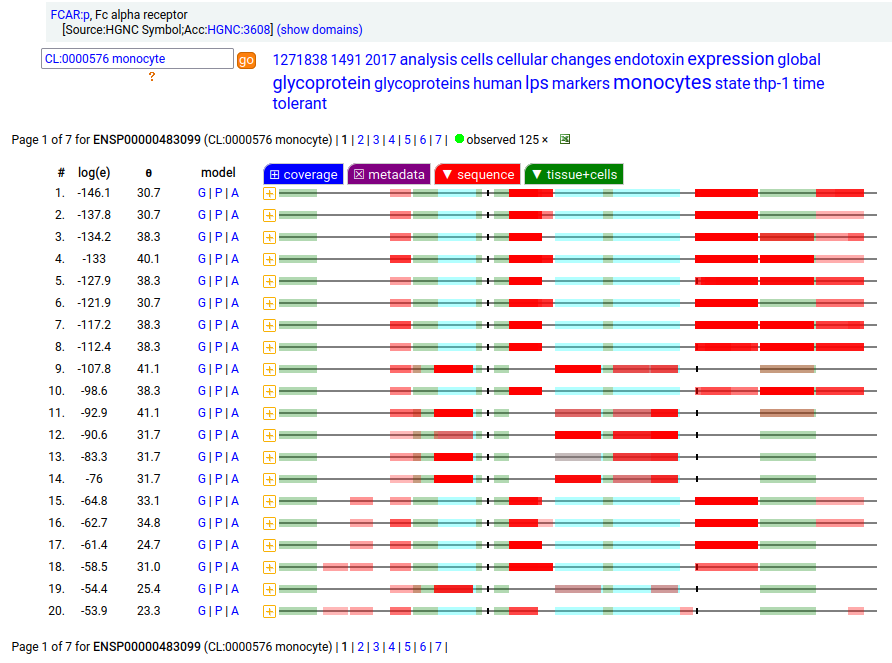
CD84 may be found in platelets, myeloid & lymphoid leukocytes: it is a type I membrane protein with 2 extracellular Immunoglobulin-like folds. Signal peptide: (1-21); extracellular region: (22-259); TM domain (224-246); intracellular Y-phospho-IDR (247-345). The soluble form found in urine has no signals cward of R131. Human cluster of differentiation member 84 (CD84:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations.  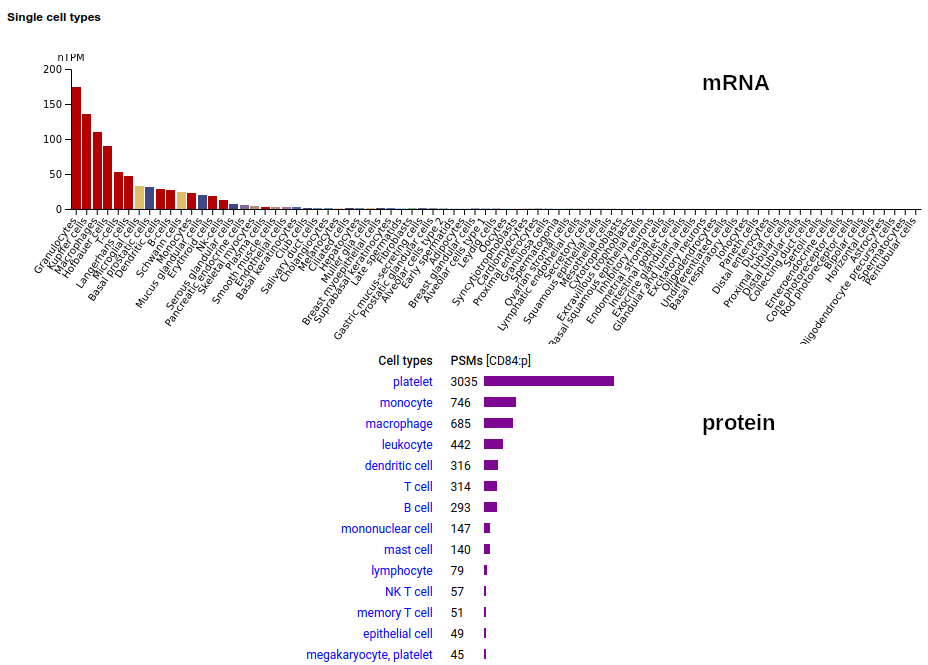
The coverage diagrams for CD84:p in "urine" vs "platelet" demonstrate the loss of the TM & intracellular regions in the soluble form. The kinases, phosphatases & proteinases that affect changes to this protein are unknown. 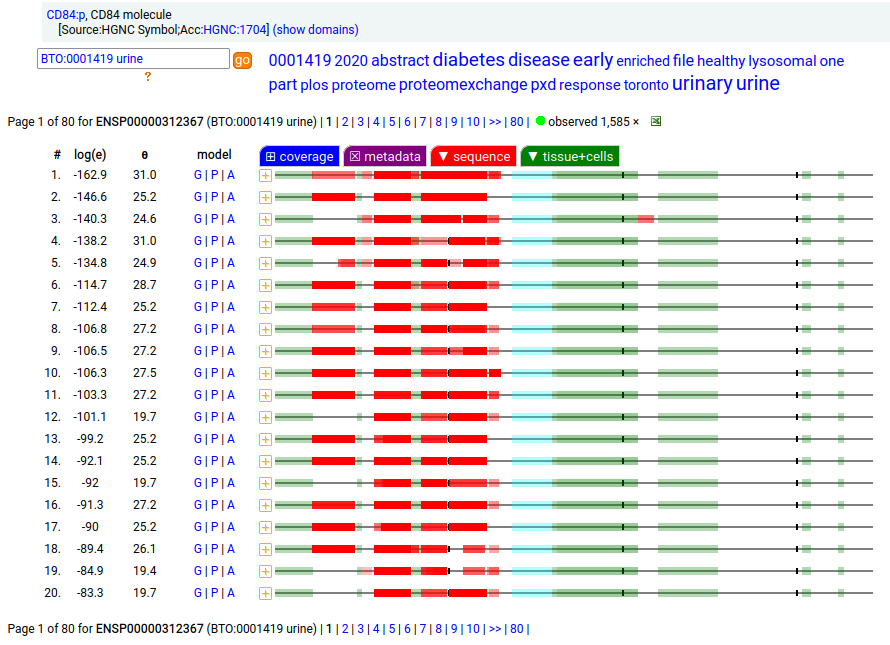 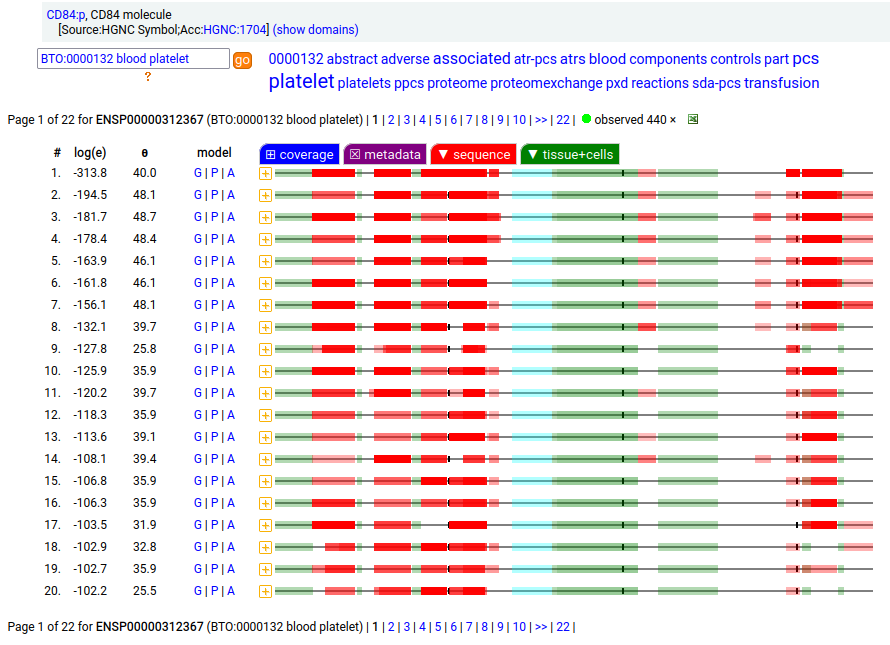
The Y-phospho-IDR in CD84:p is quite active, with at least 3 major acceptors that appear to be occupied to some extent in all leukocyte types investigated. 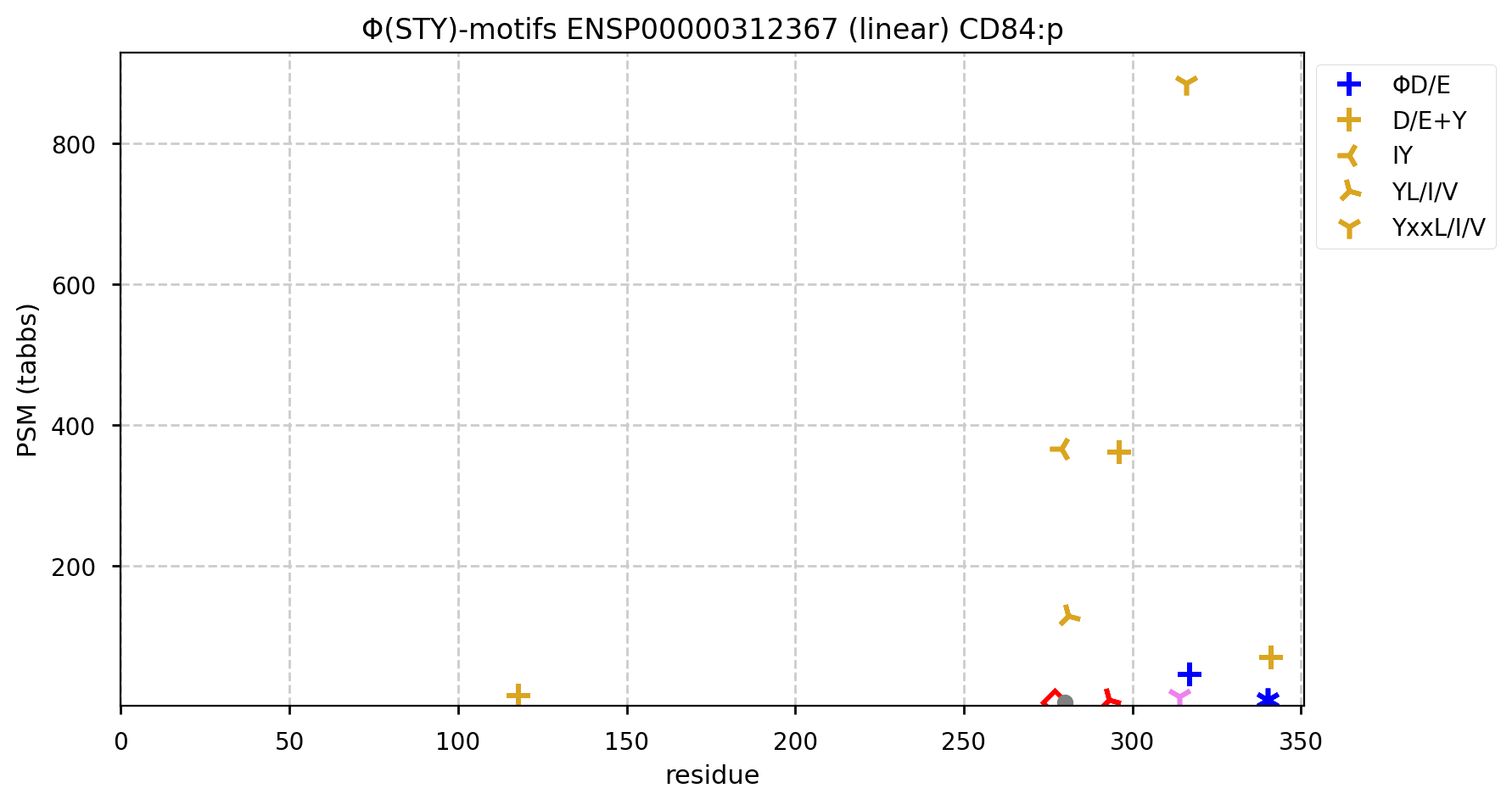
This myeloid linage cell type I membrane protein has 2 extracellular Immunoglobulin-like folds. Signal peptide: (1-17); extracellular region: (18-259); TM domain (260-282); intracellular S-phospho-IDR (283-364); N209, N230+glycosyl. The soluble form found in urine has no signals cward of R225 Human cluster of differentiation member 33 (CD33:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations. 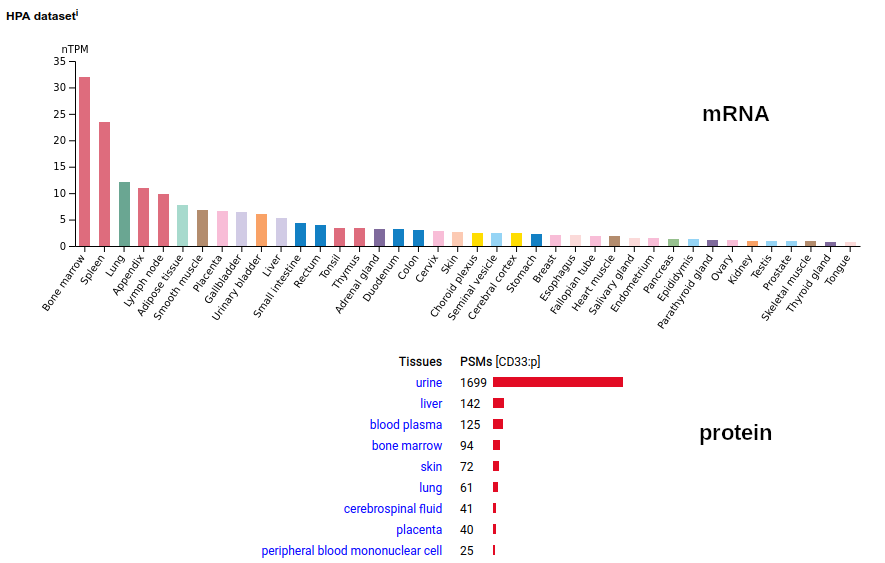 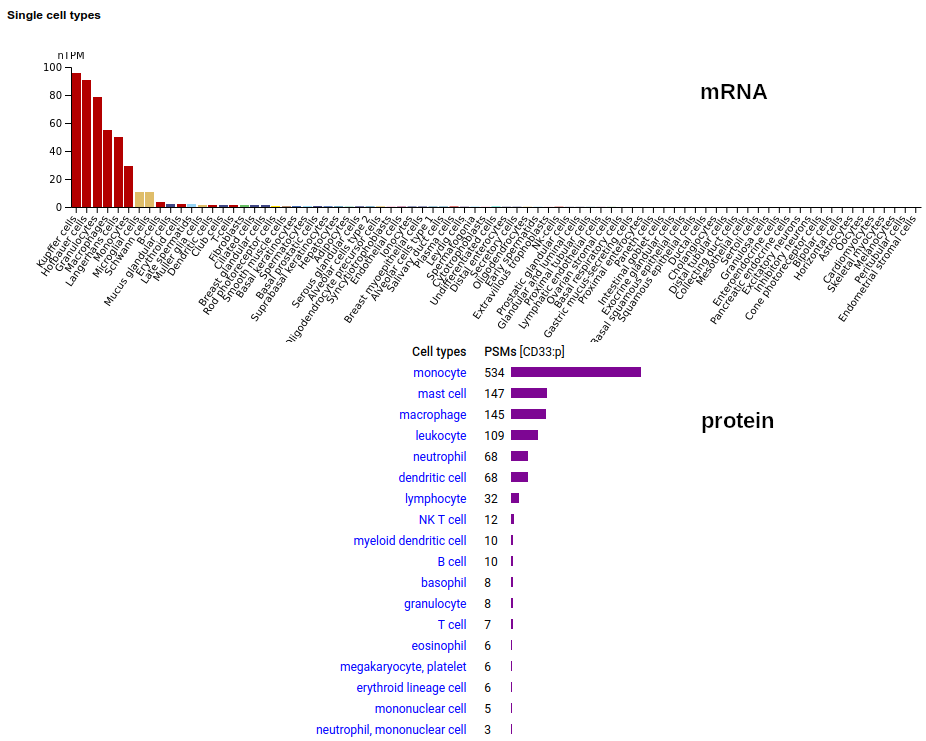
The coverage diagrams for "urine" vs "not urine" demonstrate how easy it is to detect the difference between the membrane & soluble forms using MS/MS-based proteomics. There is ongoing research about how the soluble form might be used in clinical applications, e.g. Molica 2021 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8268215/). 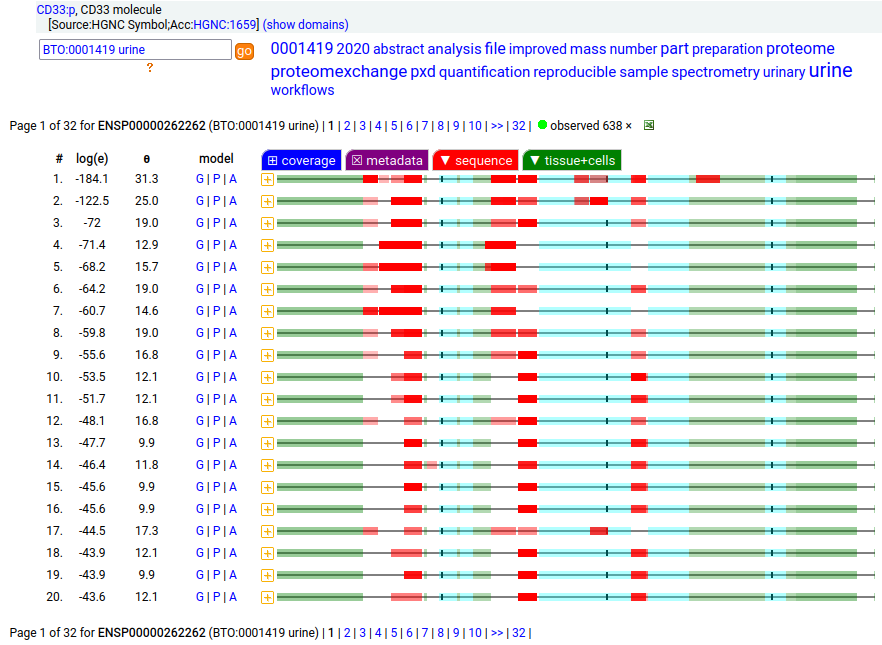 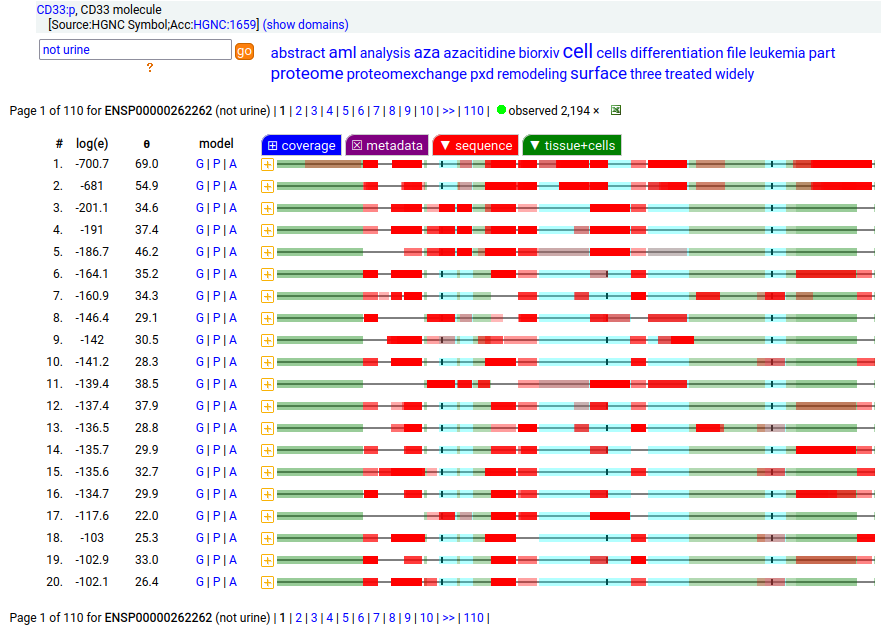
This leukocyte type I membrane protein has a extracellular TNF receptor-like fold. Signal peptide: (1-20); extracellular region: (21-385); TM domain (386-408); intracellular S-phospho-IDR (409-595); N101, N276+glycosyl. The soluble form found in urine has no signals cward of R299 Human cluster of differentiation member 30 (CD30:p, aka TNFRSF8:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations. 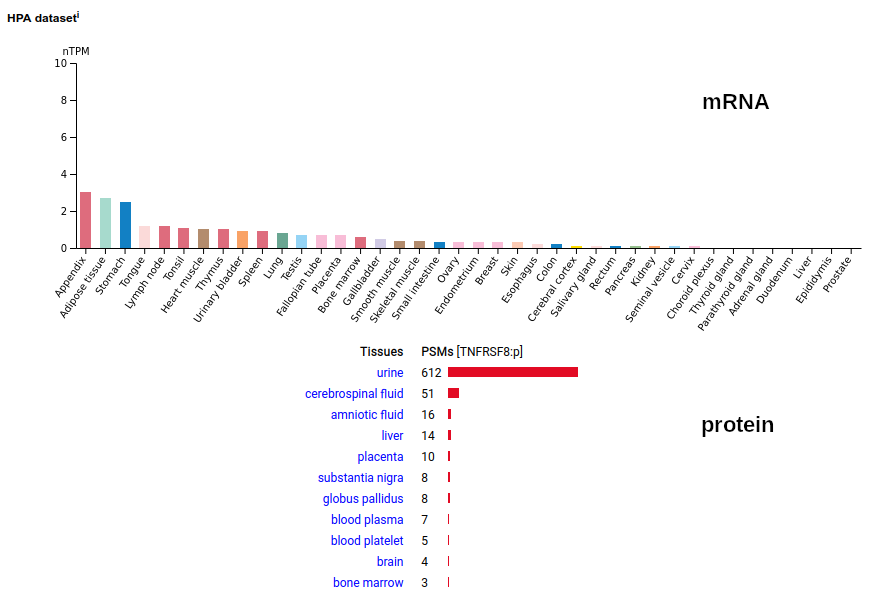 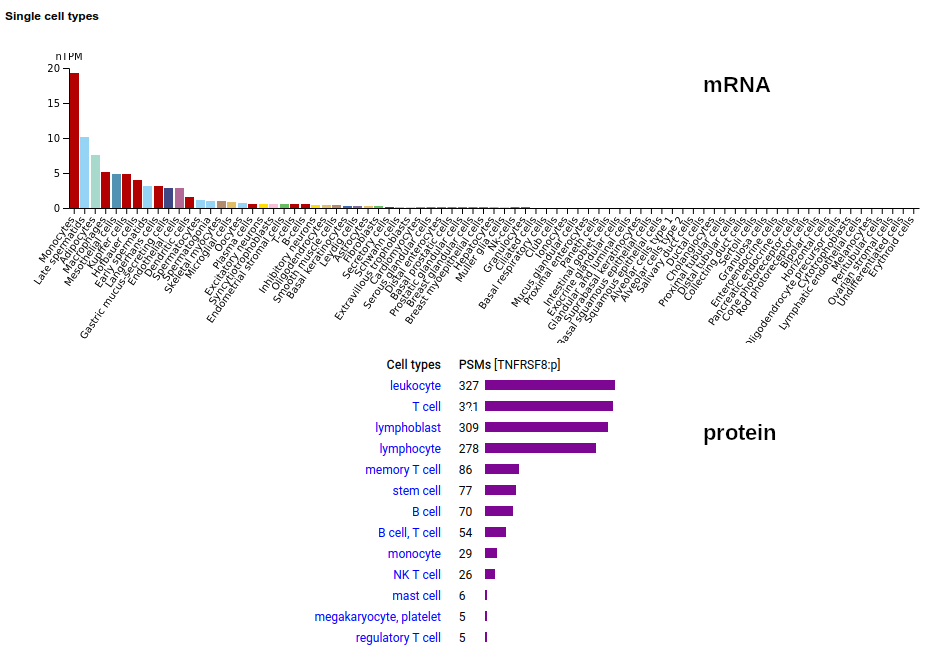
The coverage diagrams for intact, mature lymphoblast :p compared to the soluble version accessible in the urine demonstrate how easy it is to detect the difference. There has been considerable work trying to make use of the soluble form as a marker of T cell activation, e.g. Mirzakhani 2020 (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7093023/) 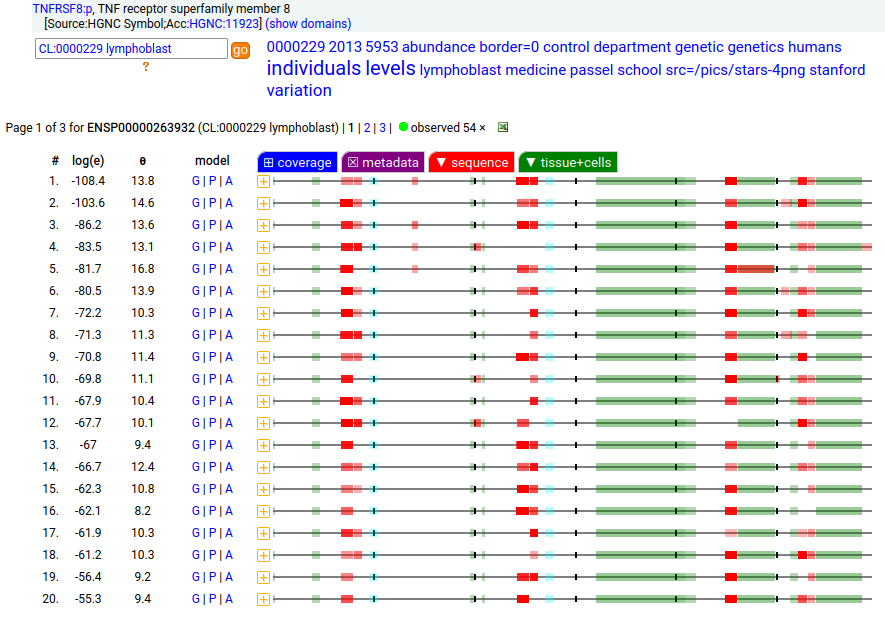 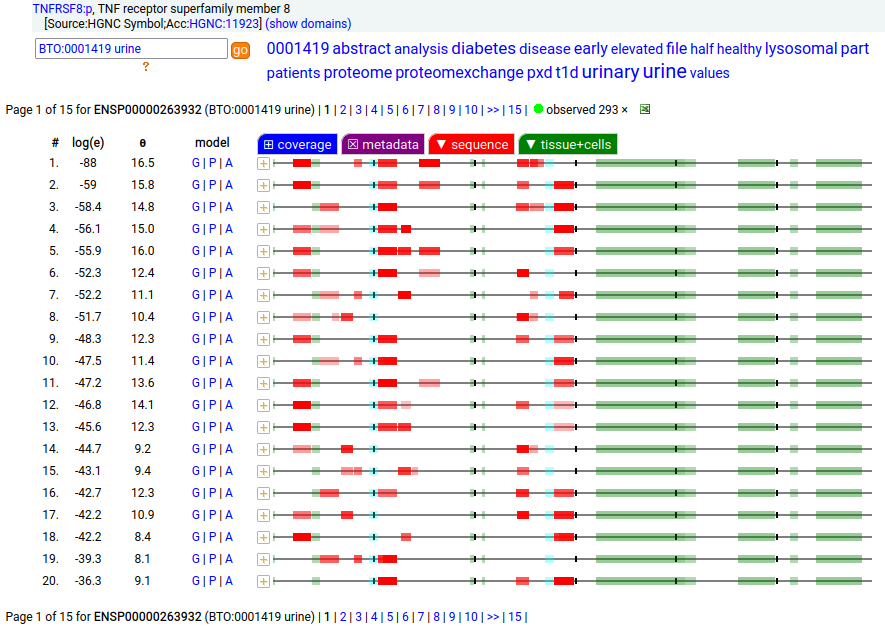
This cell surface form of this protein is found on leukocytes: it has several TNF receptor domains in its extracellular region, a TM domain (189-211) & a phosphoIDR intracellular region. The soluble form found in urine has no signal cward of R131. Human cluster of differentiation member 27 (CD27:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 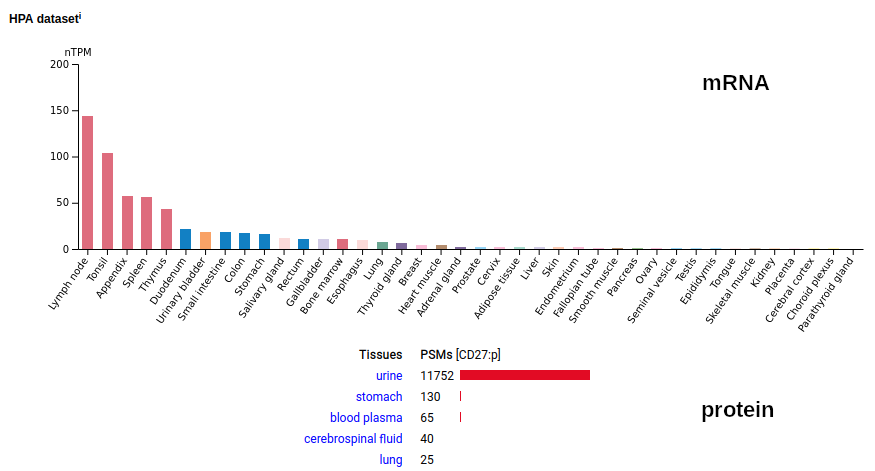
The distribution of CD27:p on leukocytes is somewhat different than the corresponding distribution of CD27:r. 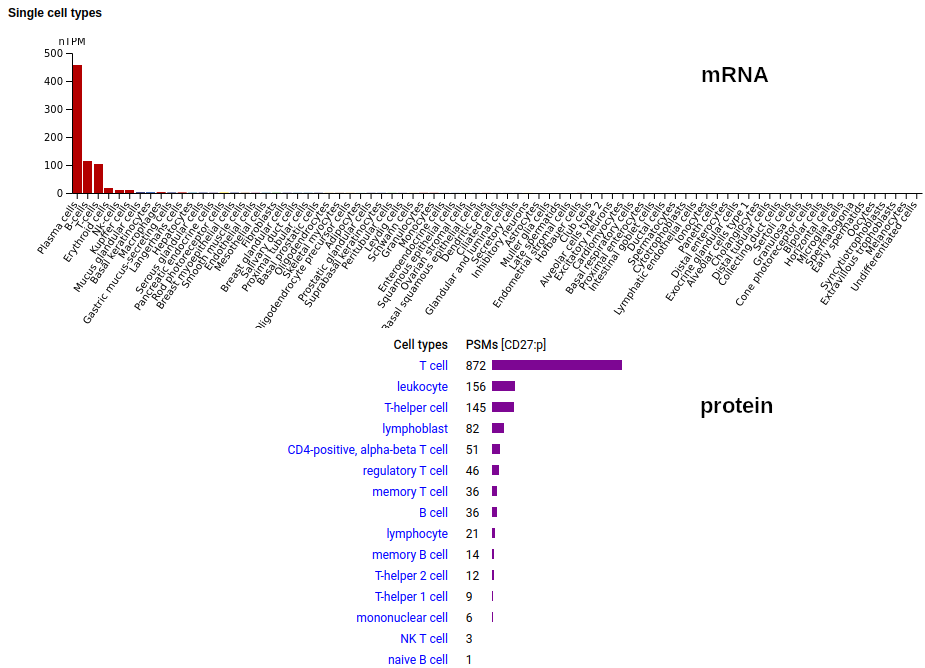
This type I membrane protein prefers to adorn T-cells, sampling the environment with a single immunoglobulin-like fold. Signal peptide: (1-25); extracellular region: (26-177); TM domain (178-200); intracellular Y-phospho-IDR (201-240). The soluble form found in urine has no signals cward of R148 & an internal cut at L130-V131. Human cluster of differentiation member 7 (CD7:p)―HPA mRNA tissue + cell type distributions & GPMDB protein tissue + cell type tabbulations. 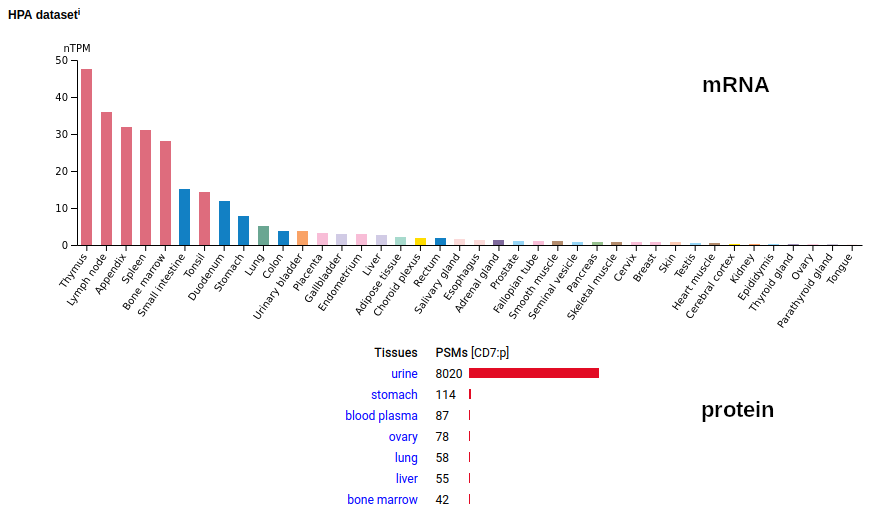 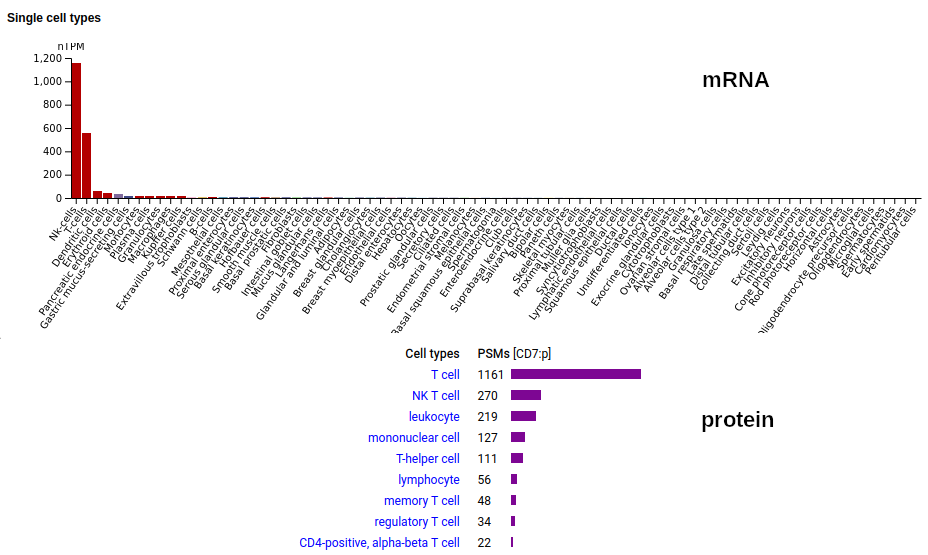
The coverage diagrams for T-cell v. urine observations of :p show a number of characteristic differences. The observations in blood plasma conform to the T-cell pattern, suggesting that the concentration of the soluble form in blood is below its LOD in typical proteomics experiments. Probably just another sad consequence of the knee-jerk application of convenient-but-poorly-thought-out FASP methods.  
This tiny little guy (25-84) is secreted, mainly by the liver, to block the efflux of iron from cells. It is processed into the peptides (60-84) or (65-84) that physically occlude the pore of the iron transport protein SLC40A1. Its antimicrobial activity is to starve bacteria of iron by removing any iron sources in surrounding tissue. Human hepcidin antimicrobial peptide (HAMP:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 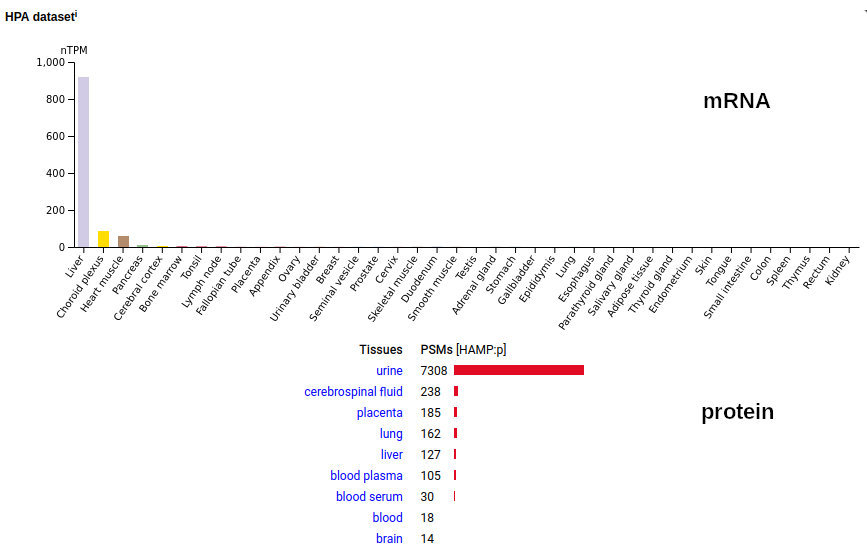
When thinking about how urine proteomics works, it is helpful to remember 2 things: 1. all blood is filtered through the kidneys about 10 times an hour; and mRNA for this protein is mainly associated with bone marrow & lung tissue, although at quite low levels. The protein seems to end up in the urine quite quickly, via transport by blood & CSF. Its mature N-terminus is D19. Human osteoclast associated Ig-like receptor (OSCAR:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 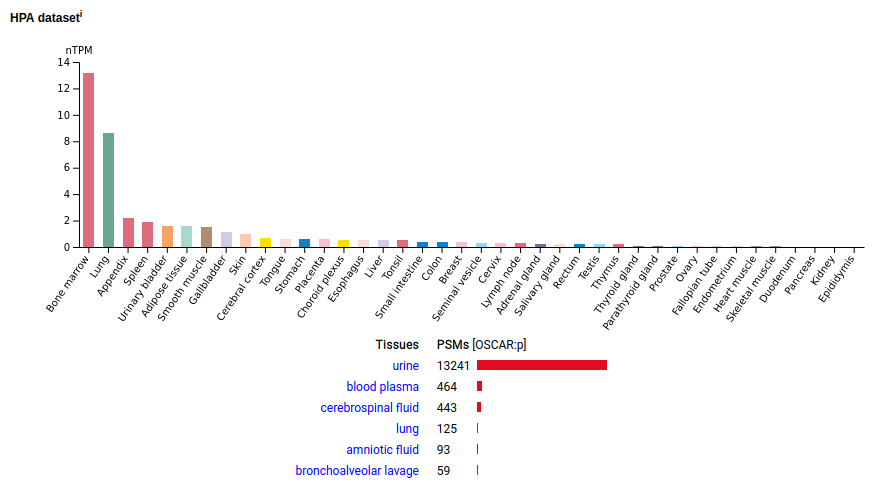
There also seems to be some story to be told about its secretion by monocyte-lineage cells, although it may be difficult to square with the oft-told osteoclast-only story. 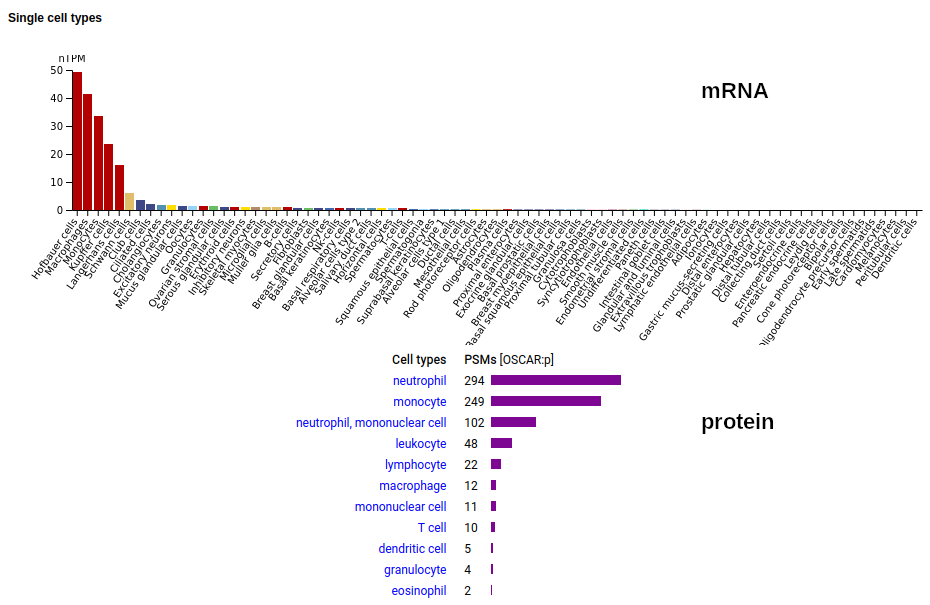
This protein is secreted, bristling with phosphorylation at >30 S/T acceptors & O-linked glycosylation at 5 T acceptors. There are 3 easily distinguished splice variants (SPP1-201, SPP1-202, SPP1-203), with the mature N-terminus for all at I17. It is the major phosphoprotein in human urine & milk where it keeps divalent cations (esp. Ca²⁺) in solution. Human secreted phosphoprotein 1 (SPP1:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 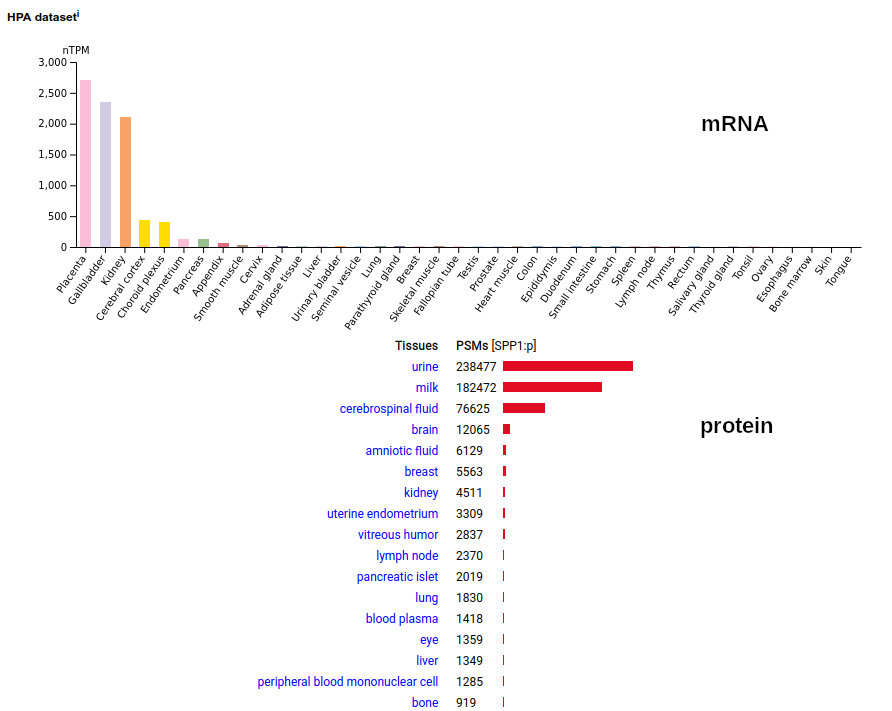
The 'zines & database blurbs have done a particularly bad job trying to come to terms with this abundant protein, probably because anything that can change local Ca²⁺ concentration may have many biological effects. Its traditional name (osteopontin) hasn't helped, as it has tethered the discussion to the tissue where it was originally described. Like most highly modified proteins, the amount of SPP1:p in MS/MS data tends to be underestimated by old-school, Seattle-style data analysis tubes. The big Kahuna of protein hormones tends to be rather illusive in proteomics data. The proprotein version of the sequence is observable in the source of the protein (pancreatic islets). In urine, the mature protein is below it's LOD; in its place the peptide removed to generate the mature hormone (57-87, C-peptide) may be observed. Human insulin (INS:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 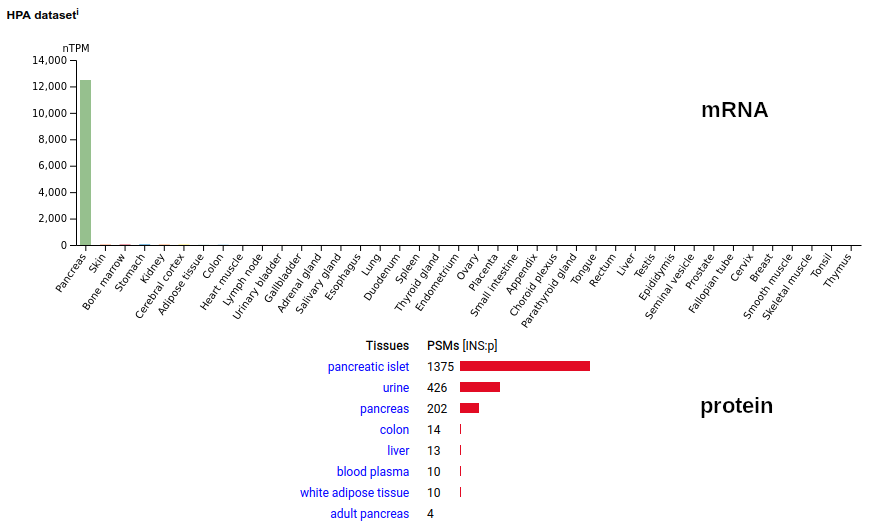
Note: the C-peptide observations in urine are from peptidome-style experiments using methods that deliberately retrieved shorter sequences. It is absent from all studies that employed any variant of the popular filter aided sample preparation idea. The rare observations of INS:p in colon and liver are the intact mature protein. The mature protein is also observed in some cell lines (e.g., HeLa), presumably from the use of recombinant human sequence added to the growth medium as a supplement.. This little guy starts out life as a type I plasma membrane protein in many tissues, with a single TM domain (169-191): this is rarely observed. The common form is caused by a proteolytic cleavage at V156 that generates a soluble protein that migrates through CSF & blood to urine. The N-terminus of both forms is either Y20 or S22. Human phosphoinositide-3-kinase interacting protein 1 (PIK3IP1:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 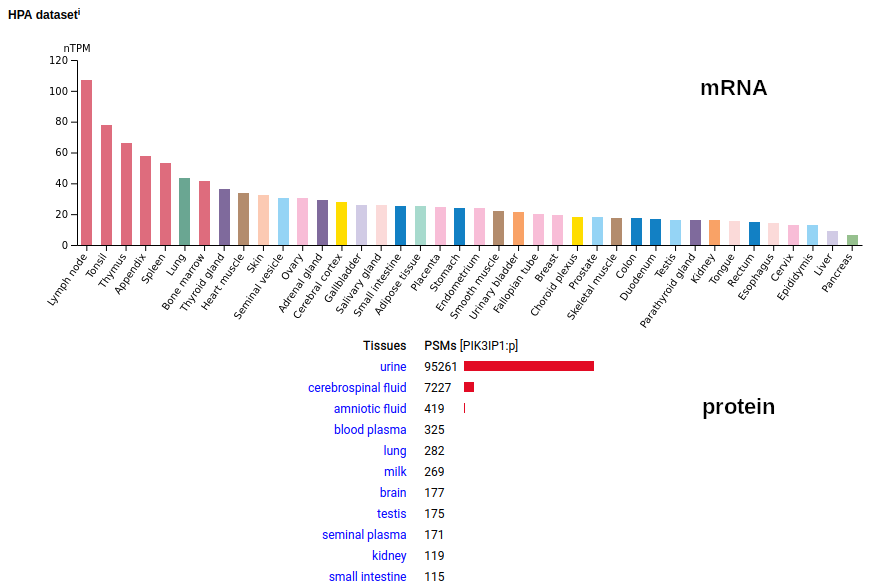
This small (260 residue) secreted protein is named for its activity: dismantling DNA chains. Extracellular DNA is an enormous PITA: the resulting sticky nets (snotballs) are weaponized by neutrophils to snare bacteria. Most of the protein's function have been examined in blood, although pee & poop serve as is its main reservoirs in humans. Mature N-terminus: L23. Human deoxyribonuclease 1 (DNASE1:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 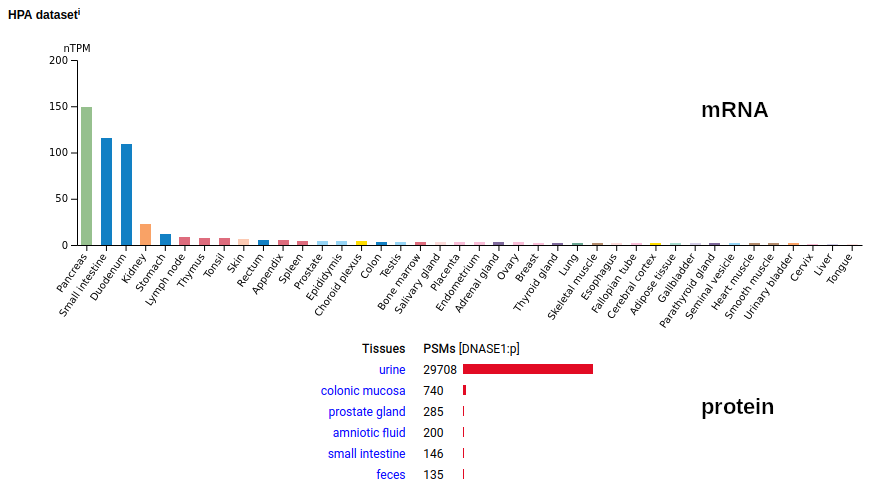
This very small (60 residue) secreted protein is named for having 3 disulphide bonds. It is produced by the stomach's epithelium and has a proposed role in the preservation of the gastric surface mucus layer. How it ends up the blood & how it is concentrated in the urine has remained a puzzle. The mature protein's N-terminus is E25. Human trefoil factor 1 (TFF1:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 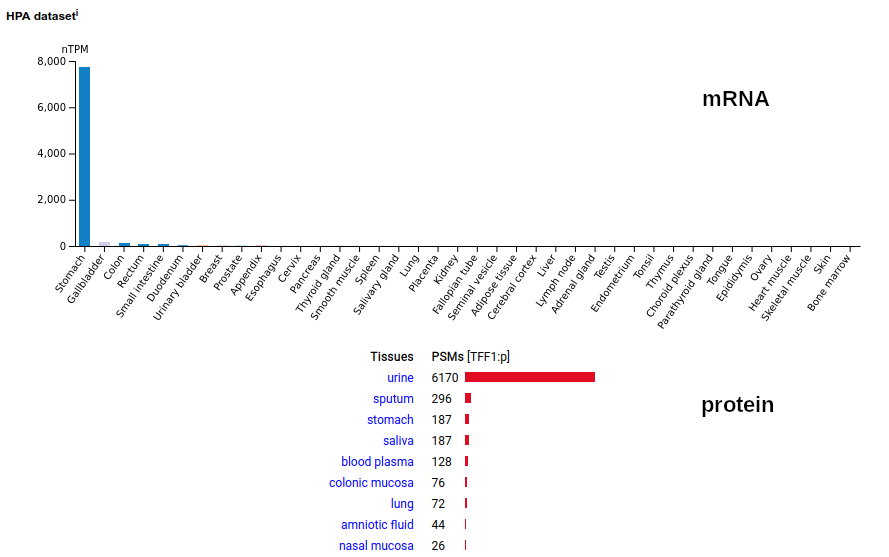
TFF1:r & TFF1:p do spike in some cancers, but the mechanism invoking them & whether they are simply an inadvertent ectopic effect or activation of some mechanism is unknown. Human trefoil factor 1 (TFF1:p)―HPA mRNA cancer tissue distributions & GPMDB protein cancer tissue tabbulations. 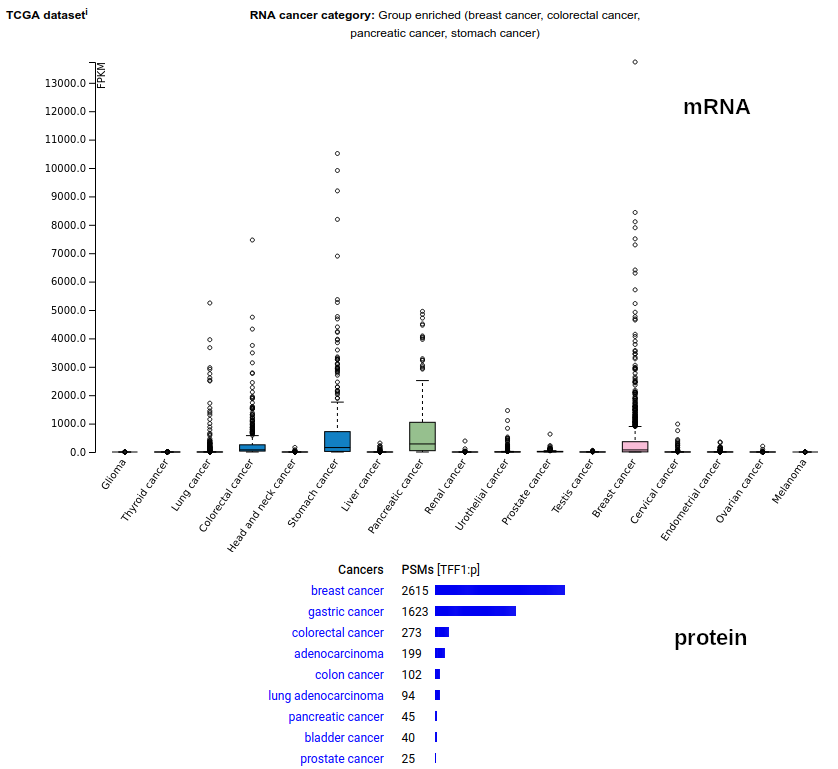
Once you get past the awful name, this small secreted protein is produced in epithelium cells in the epidermus & esophagous and sees its highest concentration in urine. It has no agreed upon function in either place, although it is not for lack of trying to find one. The mature protein's N-terminus is L23. Human secreted LY6/PLAUR domain containing 1 (SLURP1:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 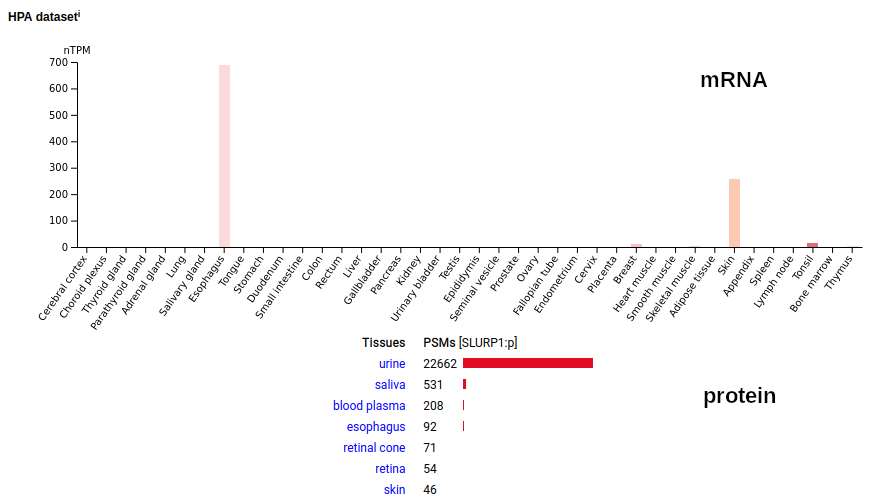
This small secreted protein is pretty much tailor-made to illustrate the importance of sources-and-sinks when trying to understand mRNA & protein tissue concentration, particularly wrt clinical applications. Mature protein N-terminus is V22. Human guanylate cyclase activator 2A (GUCA2A:p)―HPA mRNA tissue distributions & GPMDB protein tissue tabbulations. 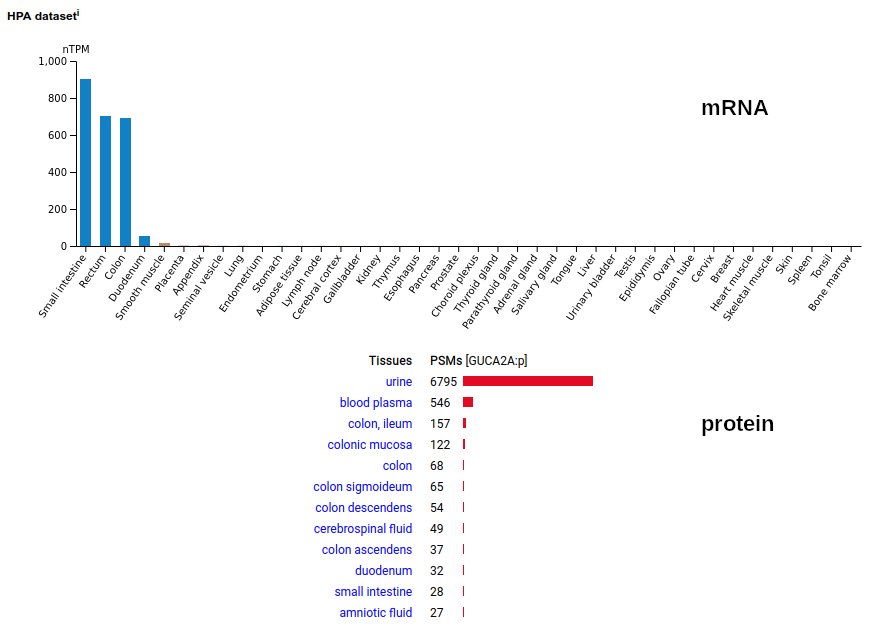
Uncoupling proteins 1-5 are mitochondrial inner membrane proteins that modulate mitochondrial function in specific tissues. Each one
has a solute carrier family 25 designation as well as a UCP gene symbol.
This protein is most frequently discussed for its role in obligate hibernator mammal brown fat. It is localized in the mitochondrial inner membrane and it functions when the animal is being roused from hibernation, shutting down ATP production in mitochondria, channeling the energy produced by the organelle into heat instead. In non-hibernating mammals its role is a little mysterious. Human & mouse uncoupling protein 1 (UCP1:p | SLC25A7:p)―GPMDB tissue tabbulations 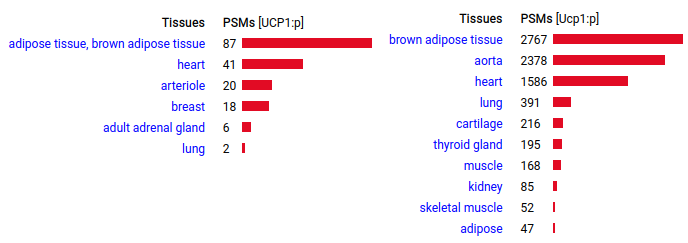
Like UCP1:p, this protein seems to be utilized more frequently by mice compared to humans, although its tissue distribution is different. The case for its role in non-shivering thermogenisis is less established than that of UCP1:p. Human & mouse uncoupling protein 2 (UCP2:p | SLC25A8:p)―GPMDB tissue tabbulations 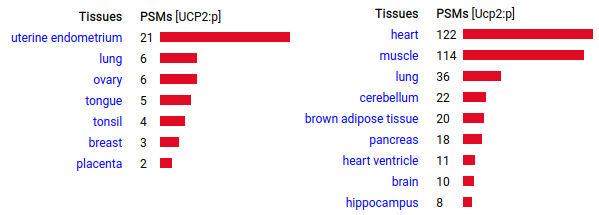
UCP3:p is also a mitochondrial inner membrane transport molecule, but it is most frequently observed in striated muscle. The idea that this sequence is used to "uncouple" ATP production to produce heat has been challenged & alternate functions proposed, e.g. Di Marchi, 2011 (https://pubmed.ncbi.nlm.nih.gov/21775425/) Human & mouse uncoupling protein 3 (UCP3:p | SLC25A9:p)―GPMDB tissue tabbulations 
The next one in the series is located in the central nervous system of both species, probably in modulating glutamate controlled proton gradients in neuron mitochondrial inner membranes. It does not appear to have any role in thermogenisis. Human & mouse uncoupling protein 4 (UCP4:p | SLC25A27:p)―GPMDB tissue tabbulations 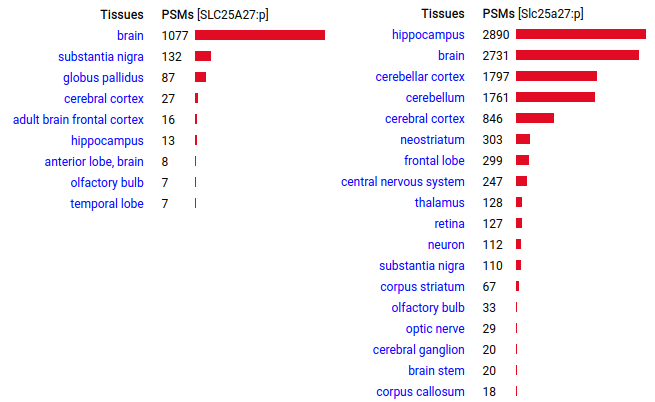
UCP5 is also found in the central nervous system of both species, with a surprise appearance in sperm/testis in humans. The role of the protein has been as a hypothetical regulator of reactive oxygen species (Ramsden 2012 https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3432969) & more recently as an anion transporter (Gorgolione 2019 https://www.sciencedirect.com/science/article/pii/S0005272819300945) Human & mouse uncoupling protein 5 (UCP5:p | SLC25A14:p)―GPMDB tissue tabbulations 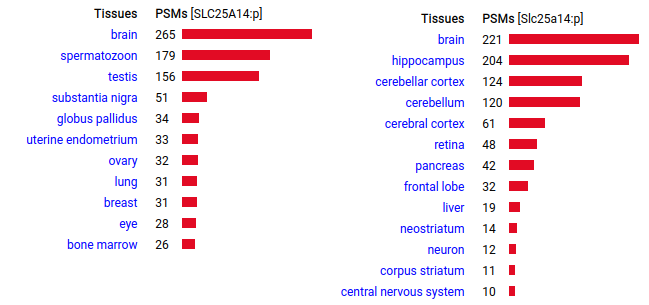
First time I noticed MHC class 2 peptides so tightly bunched around a specific structural feature (in this case a β-propeller domain, annotated on 399-659). Human low density lipoprotein receptor (LDLR:p), GPMDB coverage diagram of observed MHC 2 peptides. 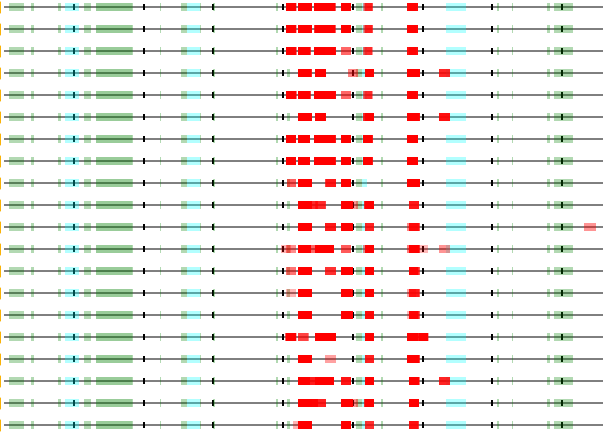
Propeller circled in red. AF model of P01130. 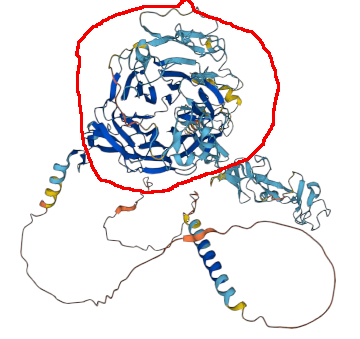
Same thing for the presented HLA class II peptides of several of the other lipoprotein receptors that have β-propellers in their structures, e.g. Human very low density lipoprotein receptor (VLDLR), GPMDB coverage diagram of observed MHC 2 peptides. 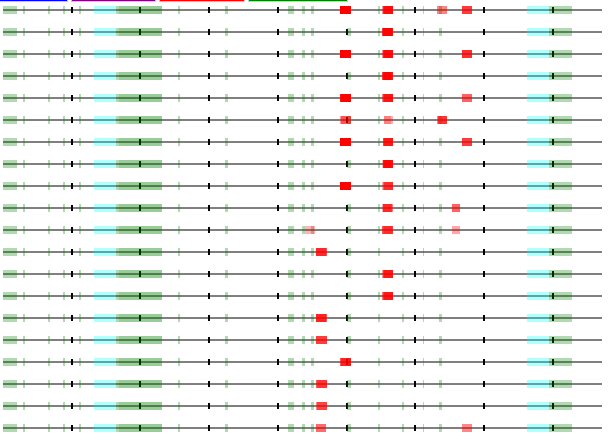
The VLDLR:p MHC 2 peptides come from the propeller in the red circle. AF model of P98155 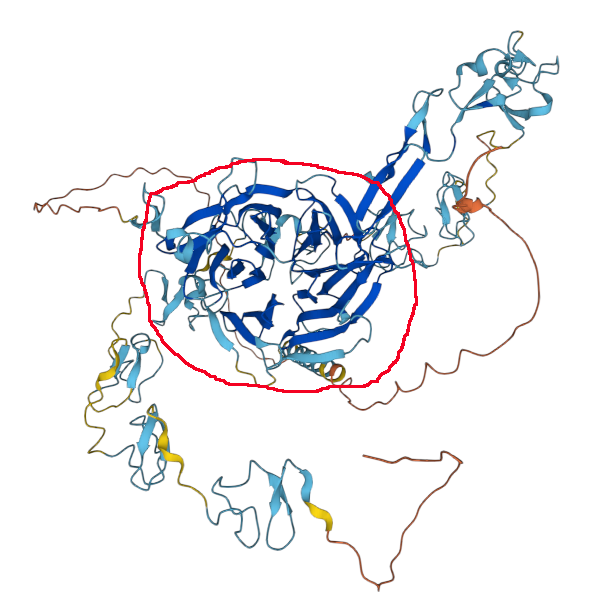
And another case where these peptides are selected from a single β-propeller domain (in red circle) within a pretty complex structure. LDL receptor related protein 4 (LRP4), GPMDB coverage diagram of observed MHC class 2 peptides & AF model of O75096.  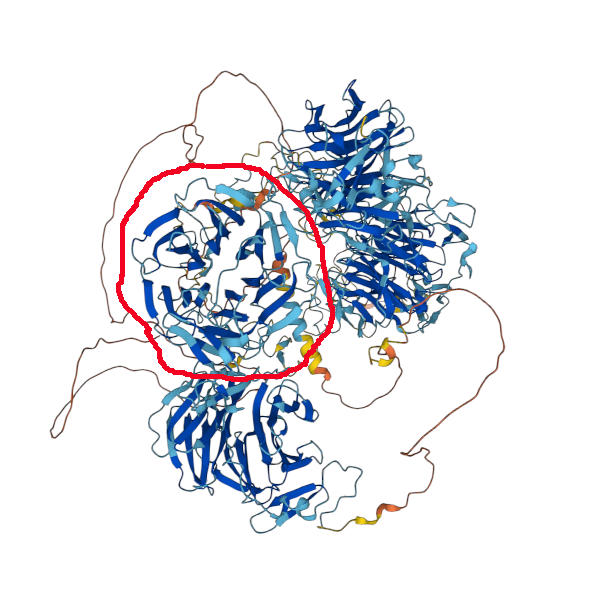
A logistic model for protein-centric PSM collections from MS/MS proteomics data. 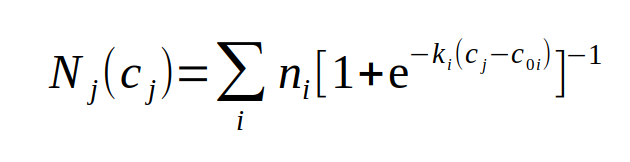
Assuming a set of peptide sequences (p) derived from a protein sequence (P), i.e., {Pⱼ | pᵢ} This model can give rise to many types of PSM v concentration curves depending on the number of peptides & the parameter values chosen. The 1st curve is for a single peptide with up to nᵢ = 10, while the 2nd corresponds to 10 peptides with nᵢ = 5 & evenly spread c₀ᵢ values. 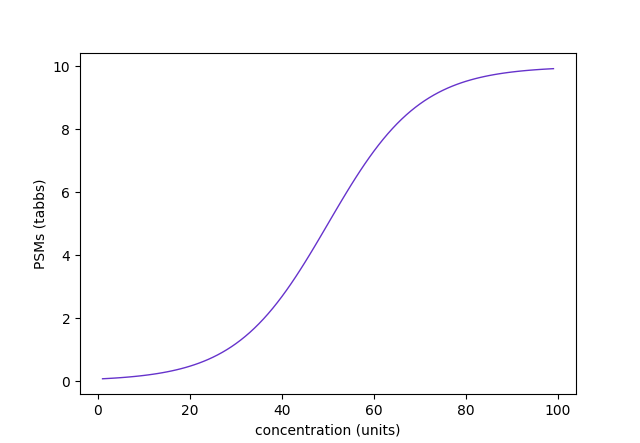 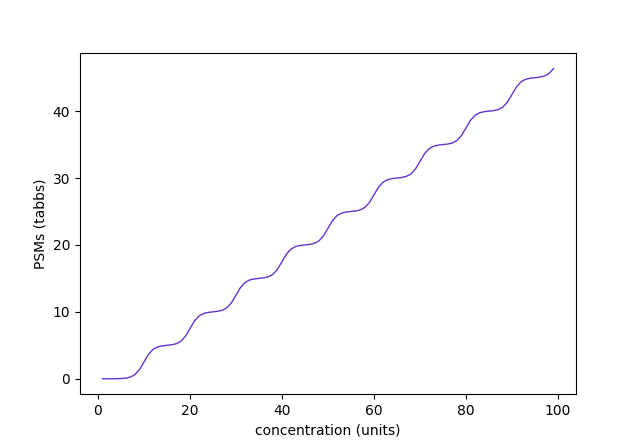
If you would like to monitor the overall level of ubiquitination in an experiment, but you don't want to go to the trouble & strife of an enrichment experiment, you can get a good feel for the overall level of this PTM by just checking for it on any one of the ubiquitin-containing genes, e.g., RPS27A:p. 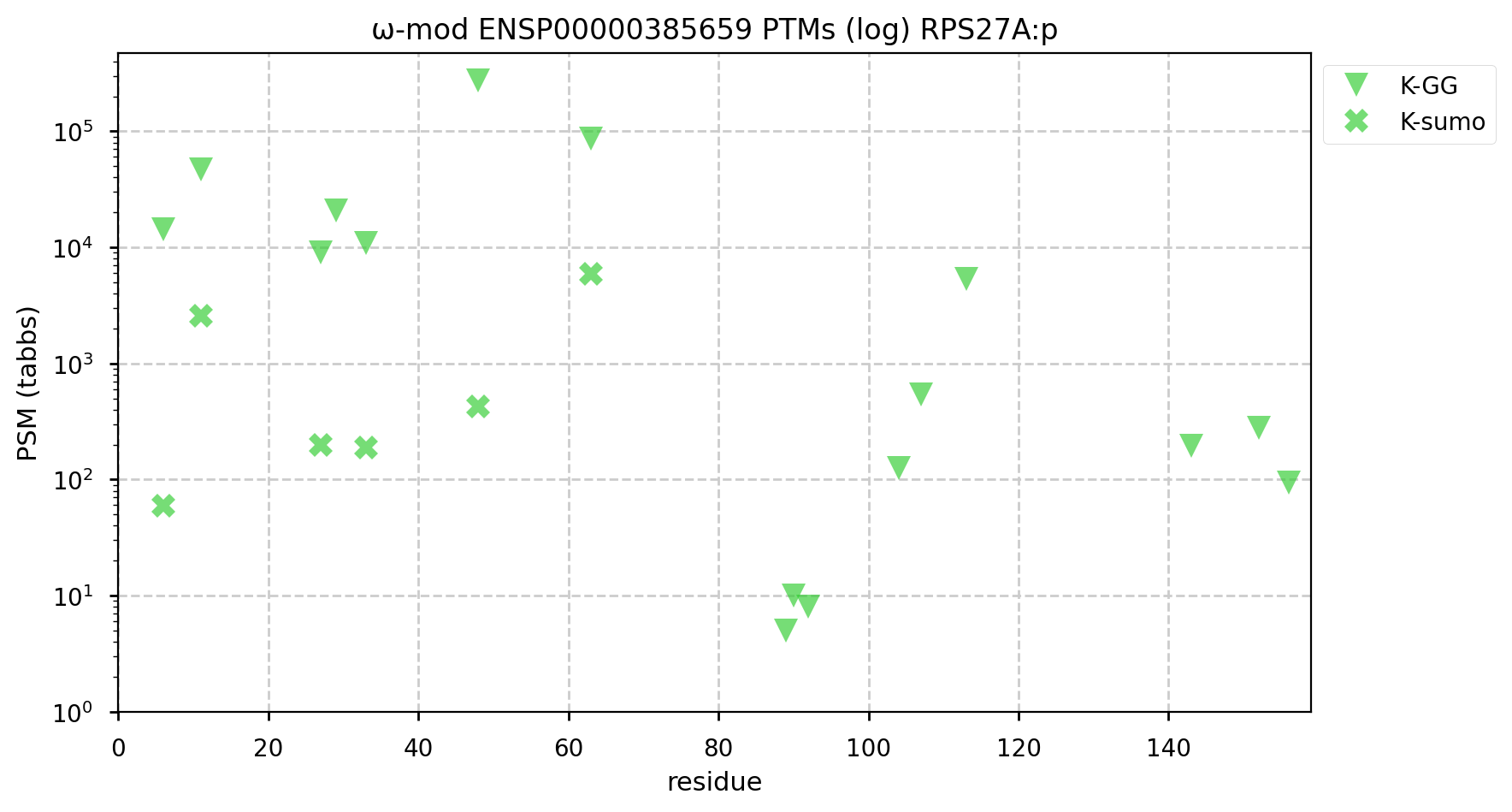
This monitoring method takes advantage of the tendency of ubiquitin to seed its own polymerization into various types of chains, triggered by its attachment to a substrate. Therefore, every Ub modified protein will generate multiple Ub-Ub crosslinks, amplifying very small, transitory individual signals into something that easily ends up over the LOD in even single shot LC/MS experiments. https://www.nature.com/articles/s41421-020-00237-y 
The Mediator complex is a large multi-subunit assembly that acts as a "general transcription factor", modulating
the activity of RNA polymerase II.
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 1 (MED1:p). Both sequences are have an nward structured domain, followed by a complex, multiple acceptor phosphoIDR that extends to the C-terminus. In addition to phosphorylation, this region features a number of interesting low complexity domains. 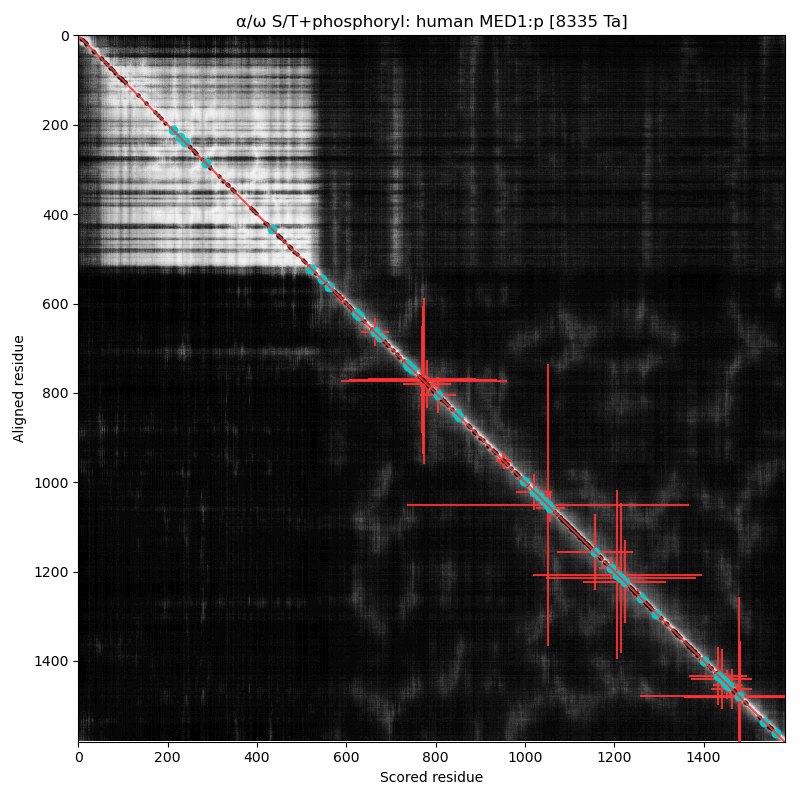 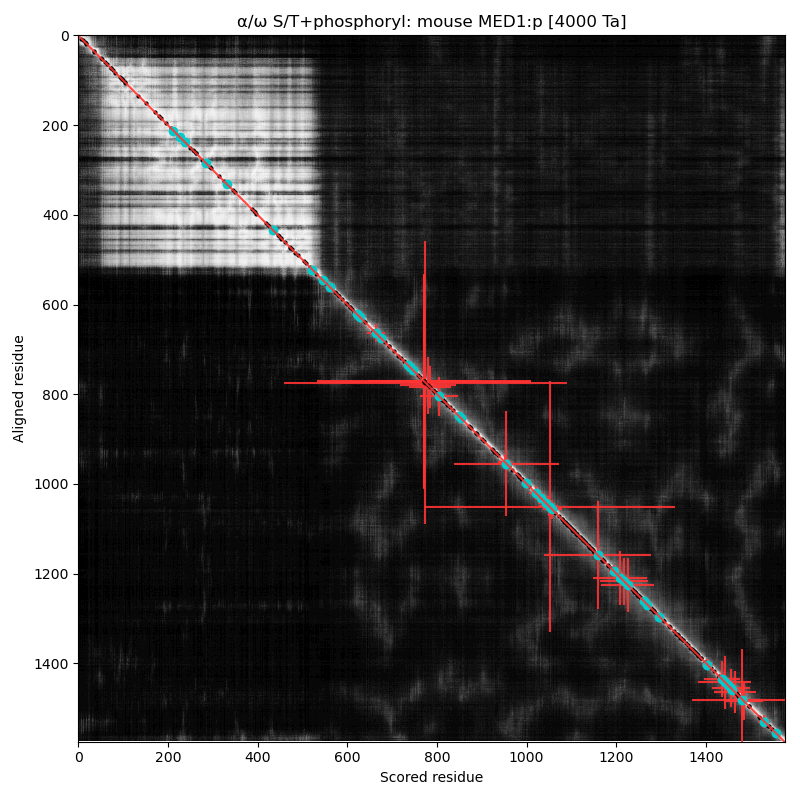
What is now :p has gone under many names in the 'zines, making the written accounts of its adventures difficult to navigate. Also known as: Activator-recruited cofactor 205 kDa component; Peroxisome proliferator-activated receptor-binding protein; Thyroid hormone receptor-associated protein complex 220 kDa component; Thyroid receptor-interacting protein; Vitamin D receptor-interacting protein complex component; & p53 regulatory protein RB18A. α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 29 (MED29:p, aka MED2:p). Unlike :p, this sequence does not use phosphorylation, even though its only has a few, scattered helical domains. Much of the information about this gene is buried in the 'zines under the subunit's older (but salacious) name "Intersex-like, IXL" (e.g., Kuuselo, 2007 https://pubmed.ncbi.nlm.nih.gov/17332321/) 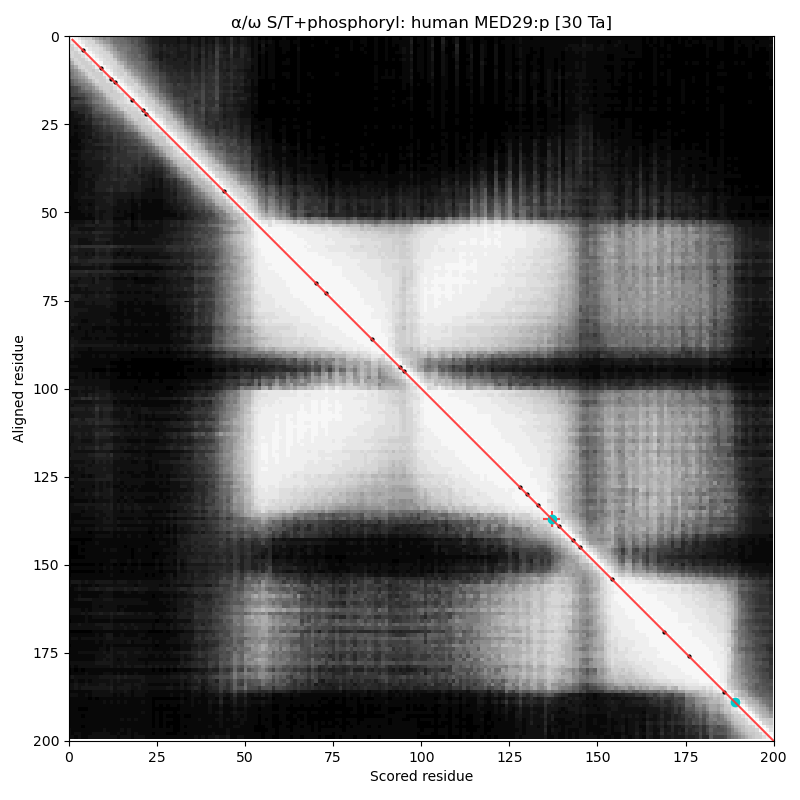 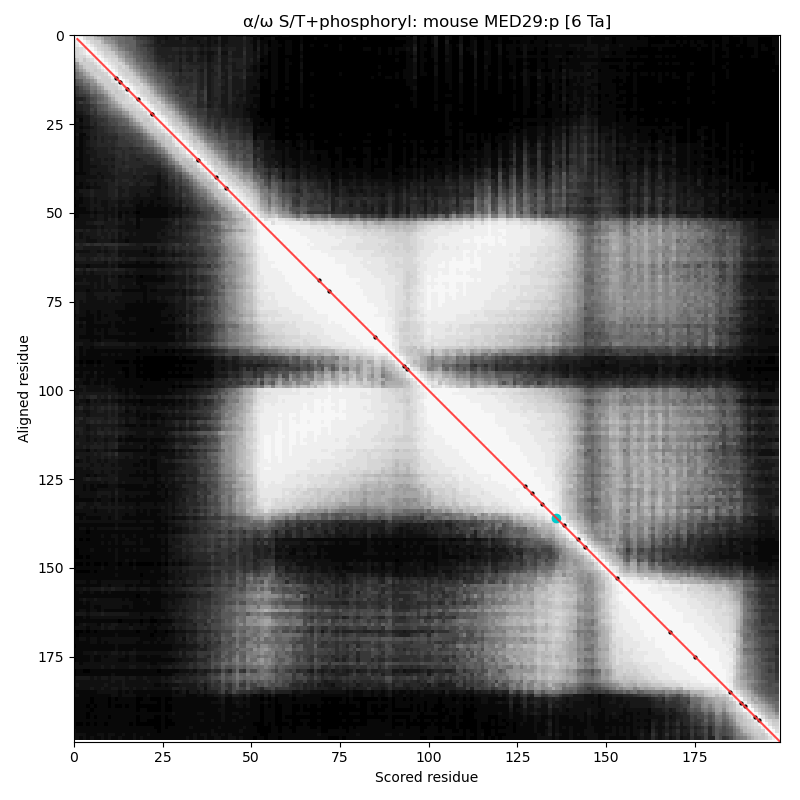
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 24 (MED24:p, aka MED5:p). This sequence's structure is a brillo-pad-like jumble of helixes, with a loopy phosphoIDR that has paired high occupancy S+phosphoryl acceptors (LxRxxΦ & ΦP) 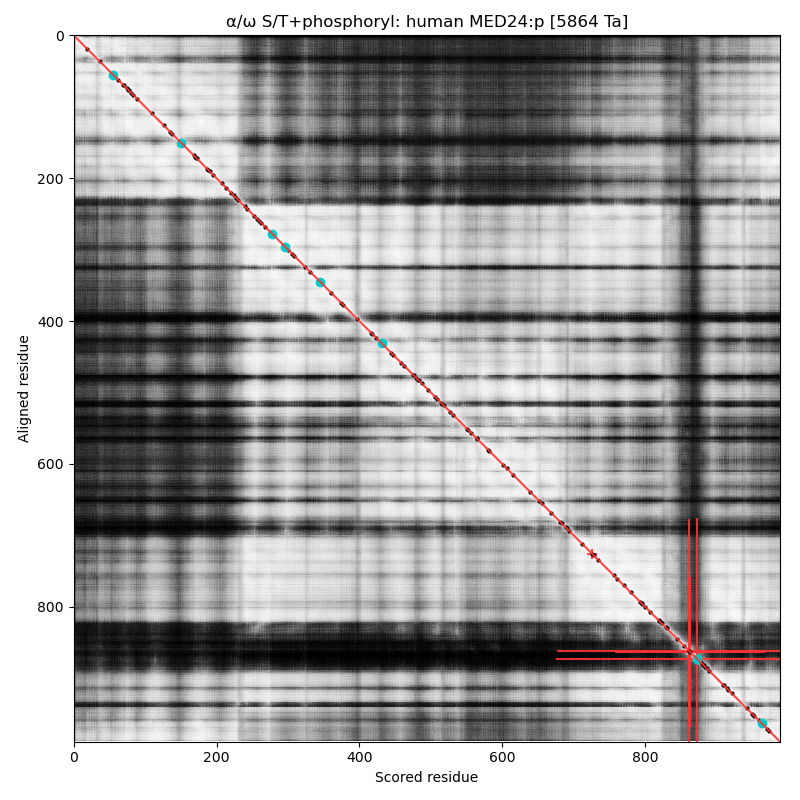 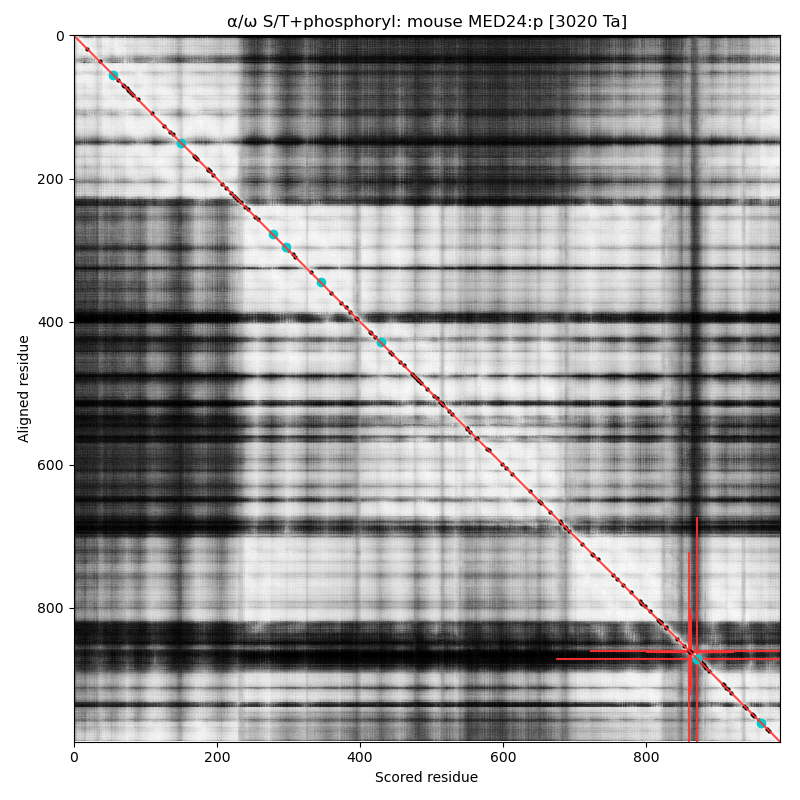
This view of the AF structure model of human has the phosphoIDR oriented so that it is sticking up on the top-centre. As with many of the Mediator complex subunits, it has been given an impressive collection of sobriquets in the 'zines: TRAP100, KIAA0130, DRIP100, CRSP100, MED5, THRAP4 & CRSP4. 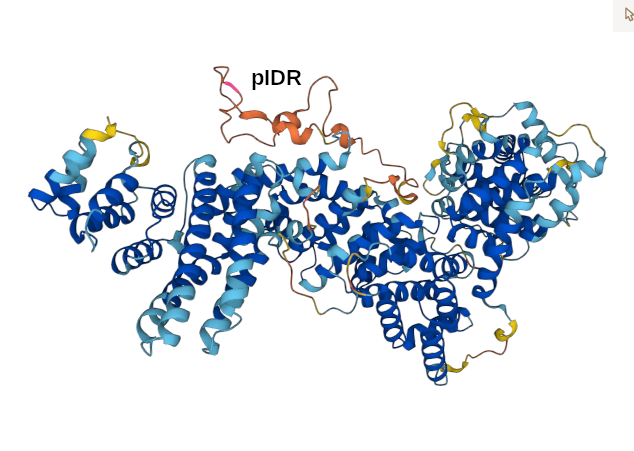
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 12 (MED12:p). This sequence's structure is a tumbleweed of helixes, with short phosphoIDRs forming fenestrations between the helical domains. It also has some low complexity regions of interest, e.g., (human 2049-2122): QQQQQQQQQQ QQQQQQQQQQ QQQQQQYHIR QQQQQQILRQ QQQQQQQQQQ QQQQQQQQQQ QQQQQHQQQQ QQQ
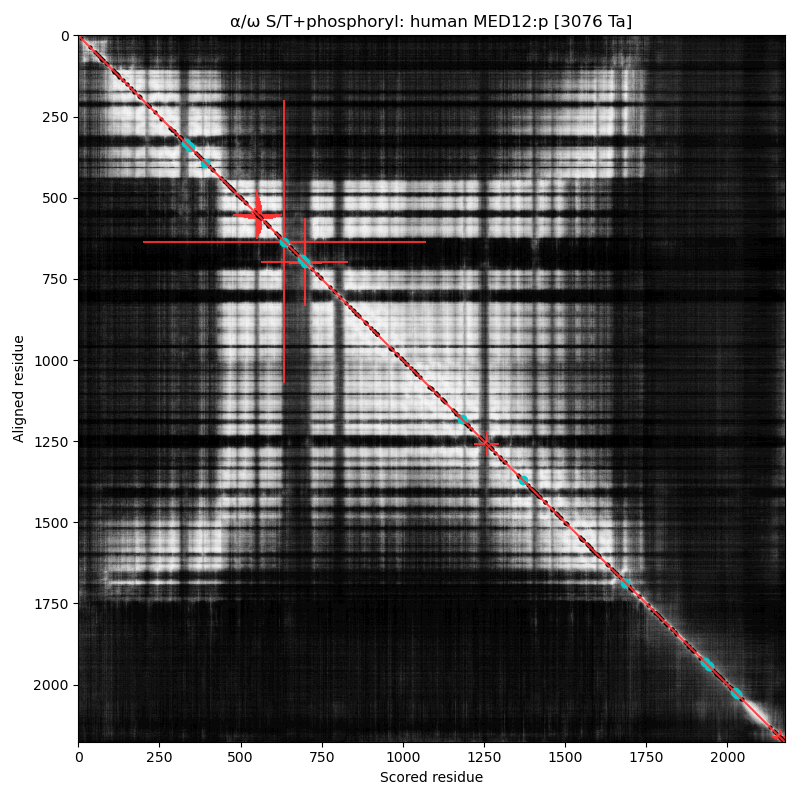 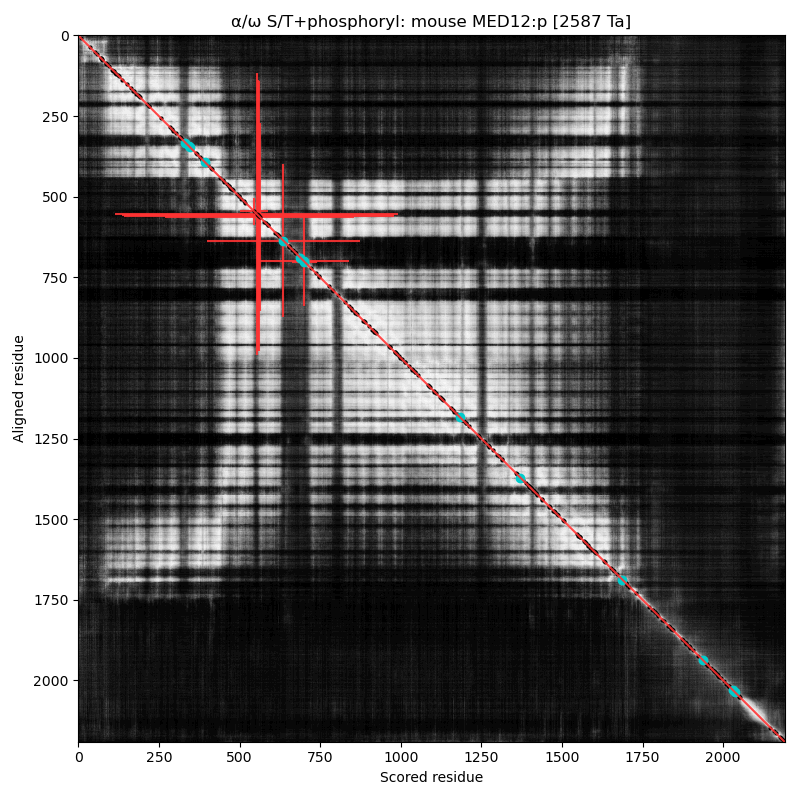
Like so many of the Mediator complex subunits, the 'zines have blessed MED12:p with a cornucopia of names & hypothetical functions: CAGH45, HOPA, OPA1, TRAP230, KIAA0192, OKS, ARC240, Kto, TNRC11 & FGS1. α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 13 (MED13:p). It has one large, multi-acceptor phosphoIDR between an N-terminal structured region & the second structured region, as well as a single acceptor phosphoIDR adjacent to a C-terminal structured domain. Also known as ARC250, DRIP250, HSPC221, THRAP1, TRAP240 & MRD61. 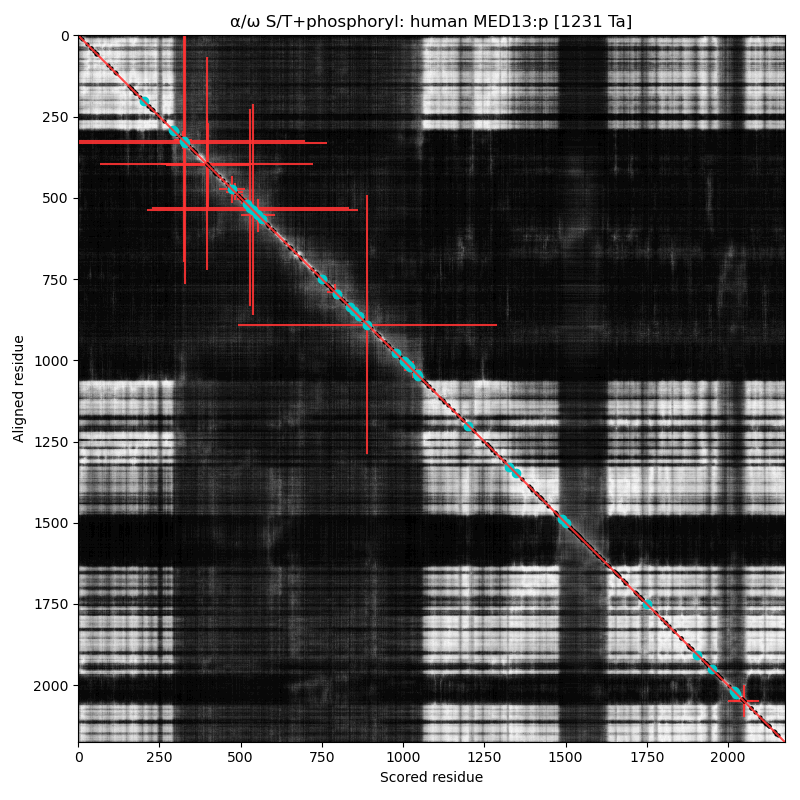 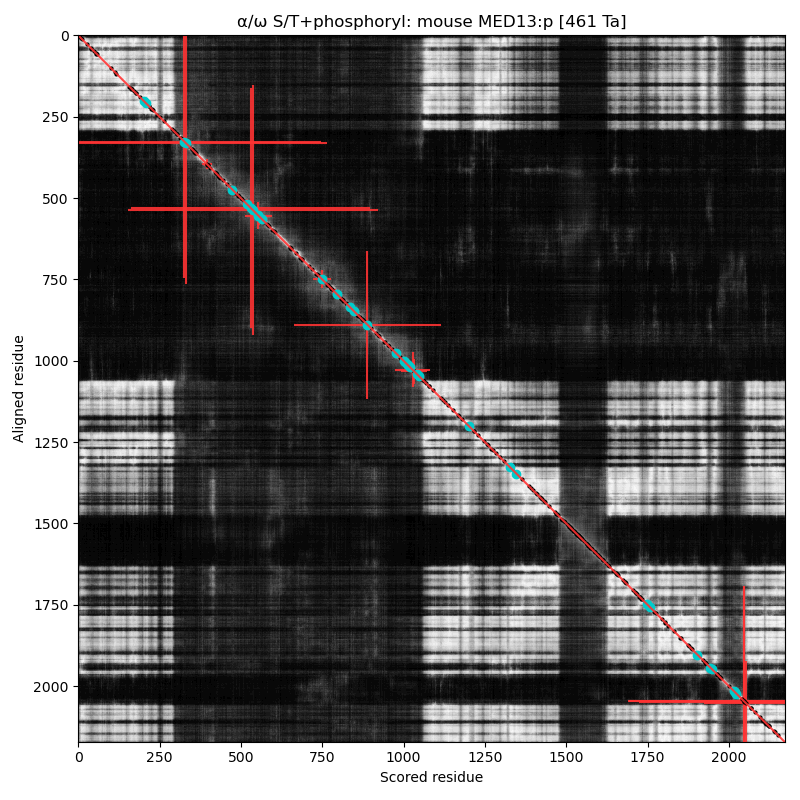
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 14 (MED14:p). The low complexity region (980-1150) with multiple S/TP motifs has several occupied acceptors, biased towards the edges of this phosphoIDR. The human acceptor S617+phosphoryl is not present in the mouse sequence. Aka EXLM1, CRSP150, TRAP170, RGR1, CSRP, CXorf4 & CRSP2. 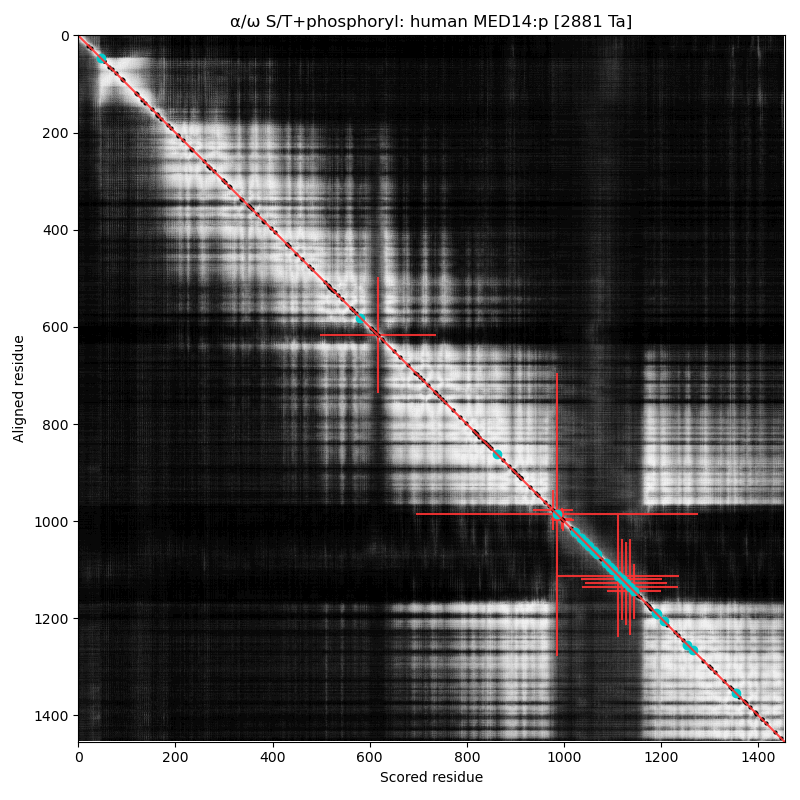 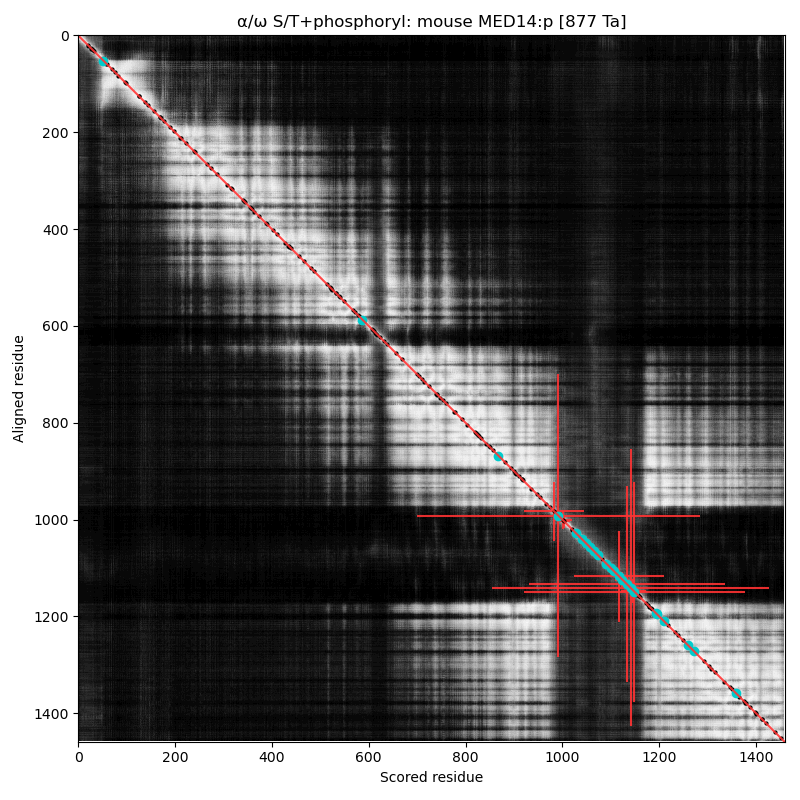
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 15 (MED15:p). S/TP type acceptors bracket one of the cward structured domains. One of the acceptors (T603 human, T604 mouse) is nearly always occupied. aka Activator-recruited cofactor 105 kDa component; Full=CTG repeat protein 7a; Positive cofactor 2 glutamine/Q-rich-associated protein; TPA-inducible gene 1 protein; Trinucleotide repeat-containing gene 7 protein 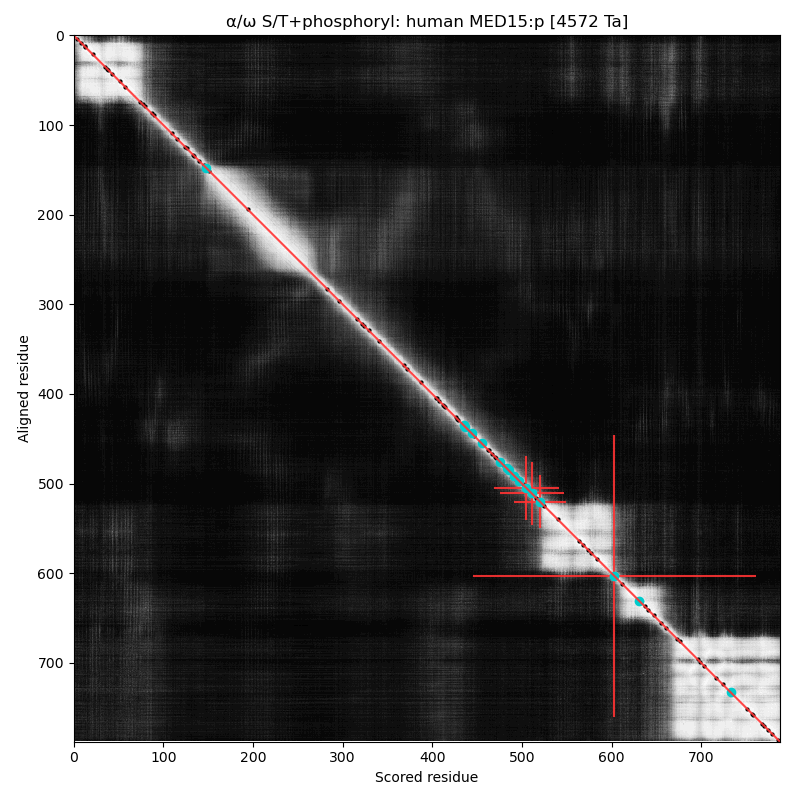 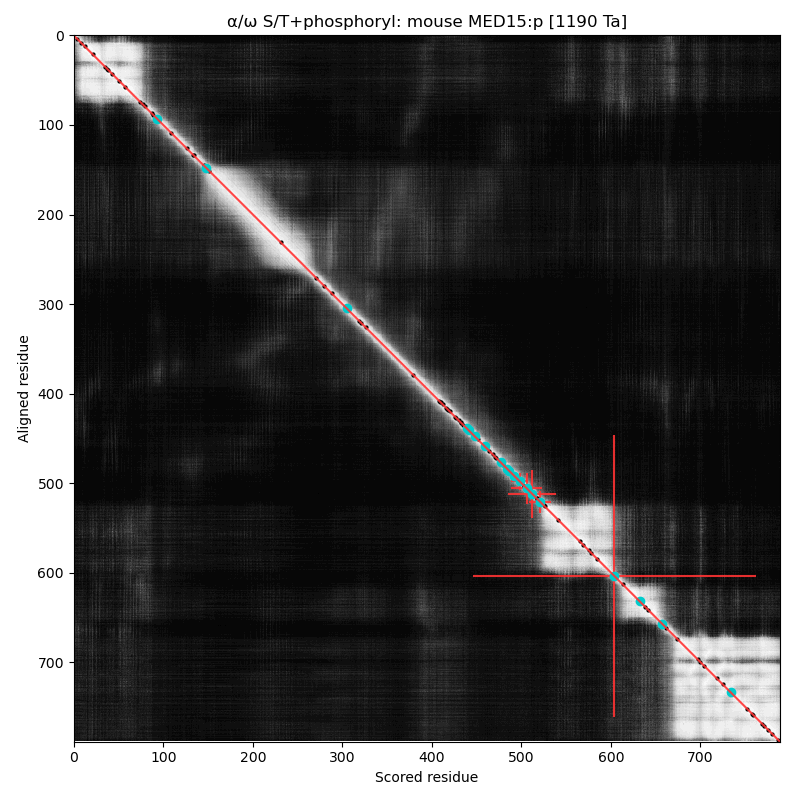
:p is one of the mediator subunits with a curious John-de-Lancie-type low complexity region (human 150-262 shown): QTQLQLQQVA LQQQQQQQQF QQQQQAALQQ QQQQQQQQQF QAQQSAMQQQ 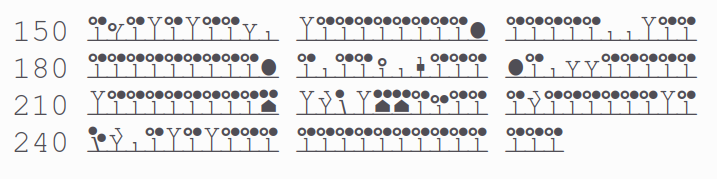
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 16 (MED16:p). The single, low occupancy acceptor (human S326) is in a narrow phosphoIDR linking domains with extended all-strand structures. Also known as Thyroid hormone receptor-associated protein 5; Thyroid hormone receptor-associated protein complex 95 kDa component; & Vitamin D3 receptor-interacting protein complex 92 kDa component. 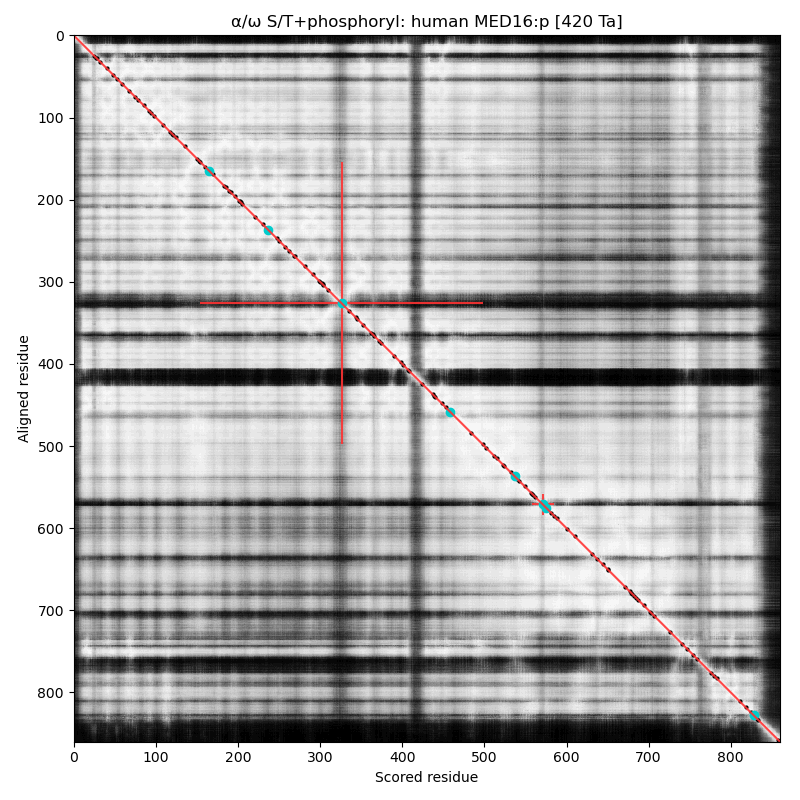 
Ubiquitination seems to be doing some heavy lifting with :p. 
α⧸ω ST phosphorylation diagrams for human & mouse mediator complex subunit 19 (MED19:p). Has a dragon's tail phosphoIDR with single high occupancy SP-type acceptor.  
Further info:
These 4 viral proteins are all observable in HEK-293T proteomics data, to various extents. The SV40 "T" protein is a deliberate addition to aid in molecular biology manipulations, while the mastadenovirus proteins were added as part of the transformation creating the original HEK-293 cell line. 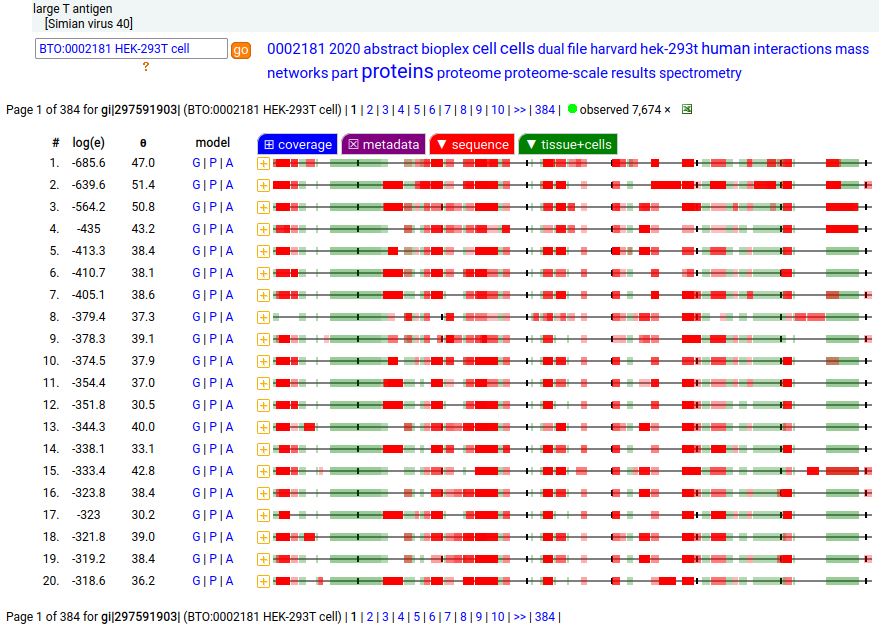 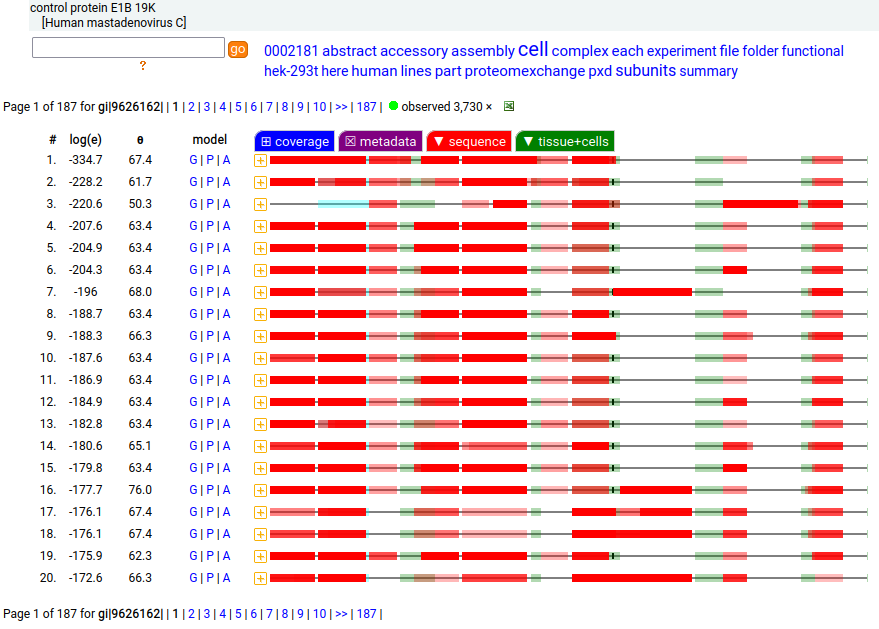 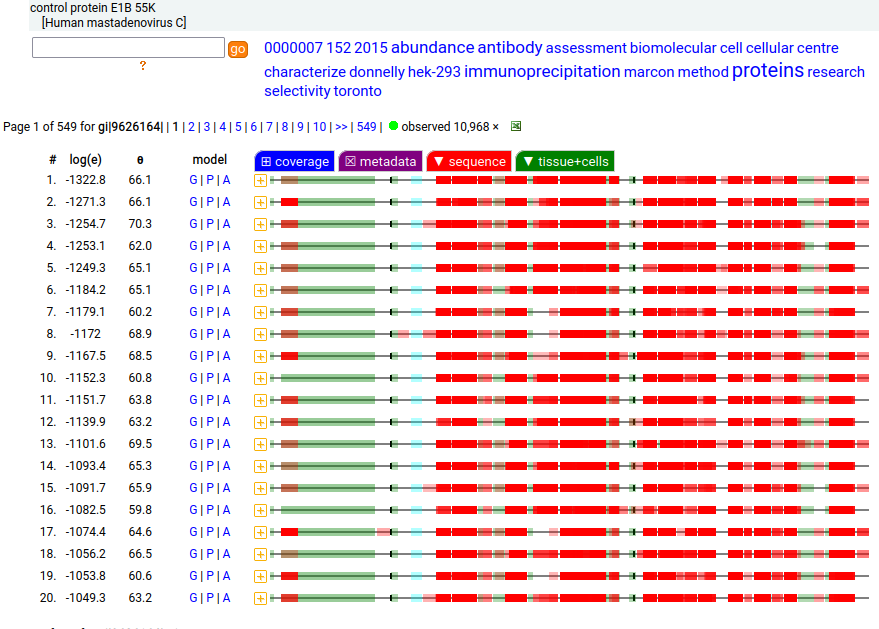 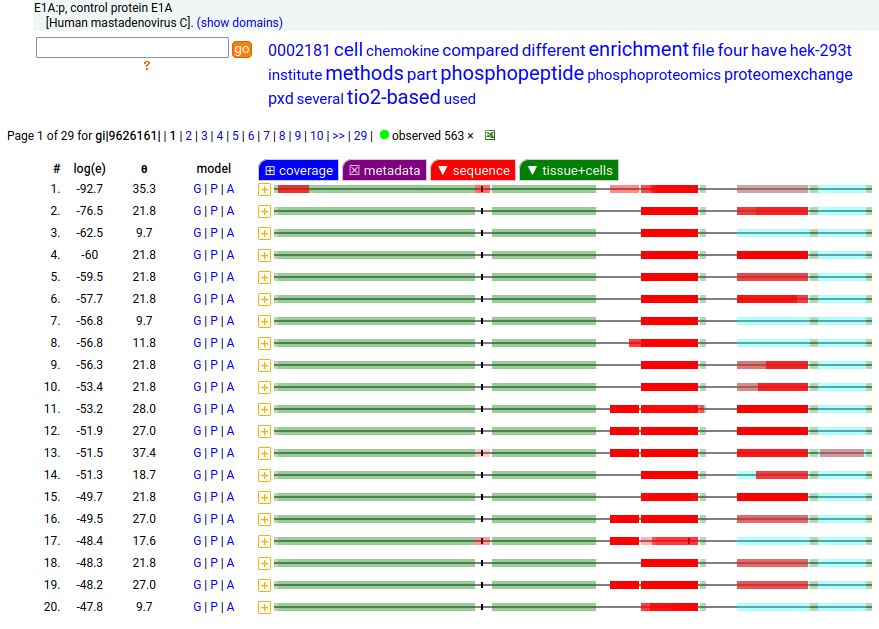
Copyright © 2024, The Global Proteome Machine.
Located at 137 Bannatyne | Privacy Statement
|


