
|
|
|
A handful of human genes have a sequence with a
translation that ends in "KDEL". These proteins end up in
the endoplasmic reticulum and they are actively returned to the ER if they
end up flowing into the Golgi apparatus. This situation is discussed below
in the form of a hypothetical Q&A session called "proteomics_101".
Genes that contain a homeobox DNA sequence can be detected
by using a regular expression on that sequence. The protein residues translated
from the homeobox DNA is referred to as a homeodomain: these domains have been
found experimentally to have a common rigid structure and bind to specific sequences
on chromosomes making them putative transcription factors of one type or another.
Homeodomain containing proteins tend to be mainly (>50%) IDRs that are
phosphorylated in evolutionarily conserved patterns. These are their stories.
A scrapbook of select human liver-specific protein Q-stickers, that has been featured on Bluesky and Mastodon.
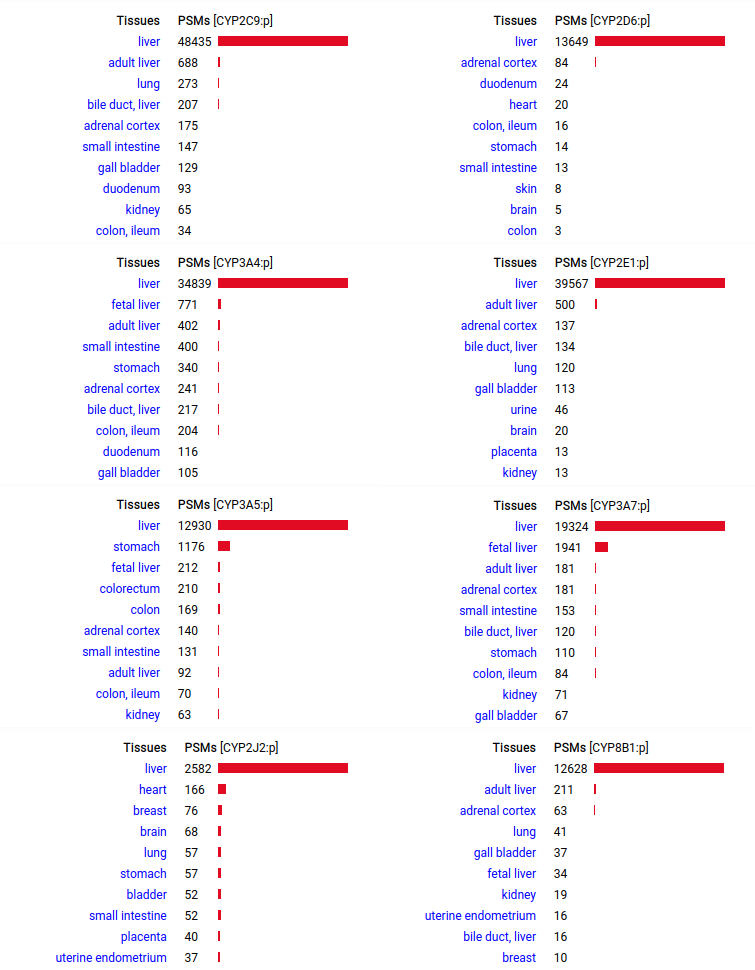
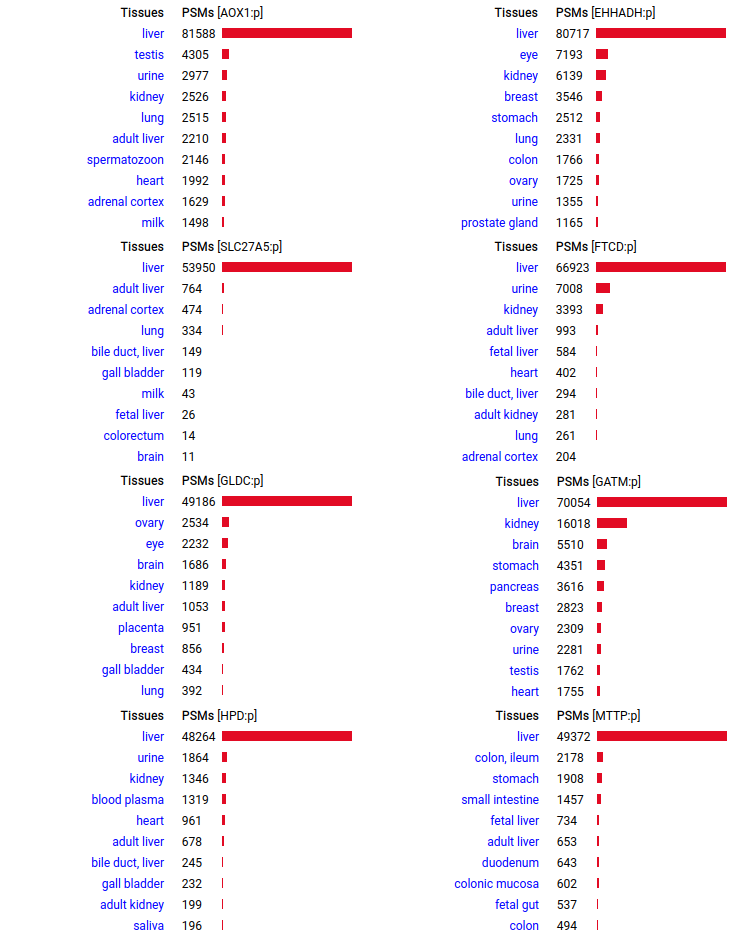
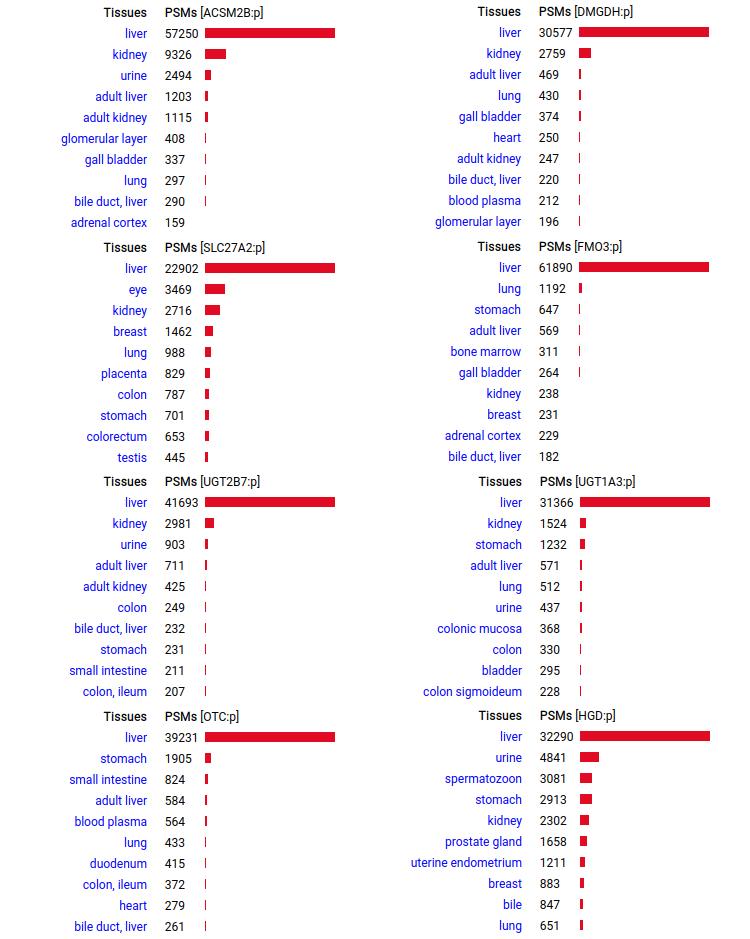
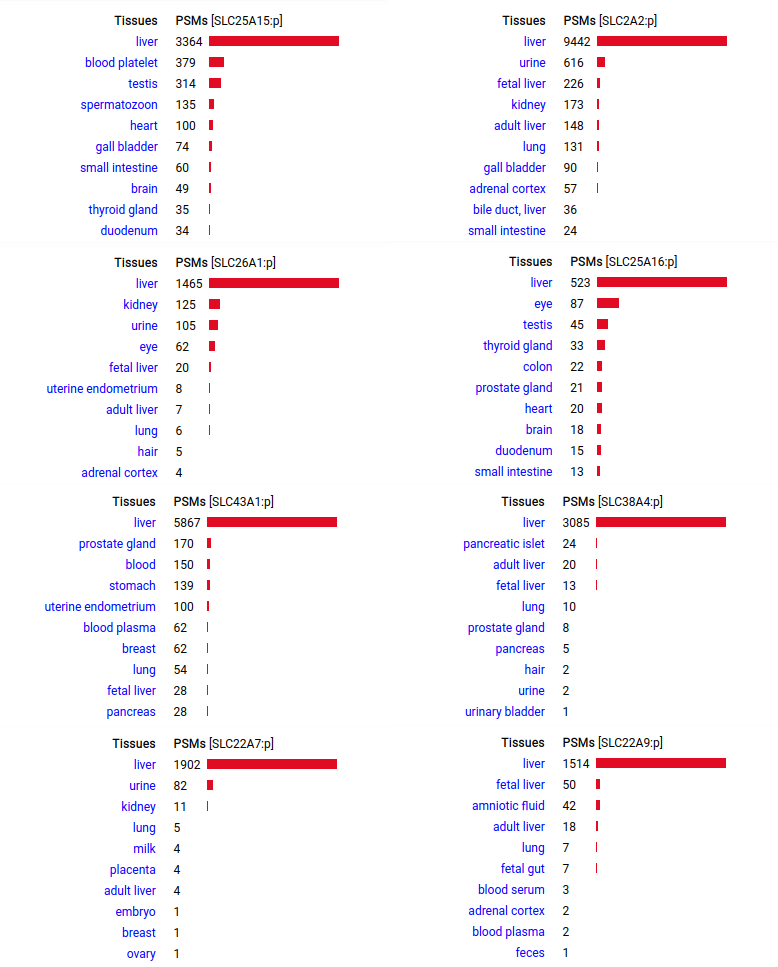
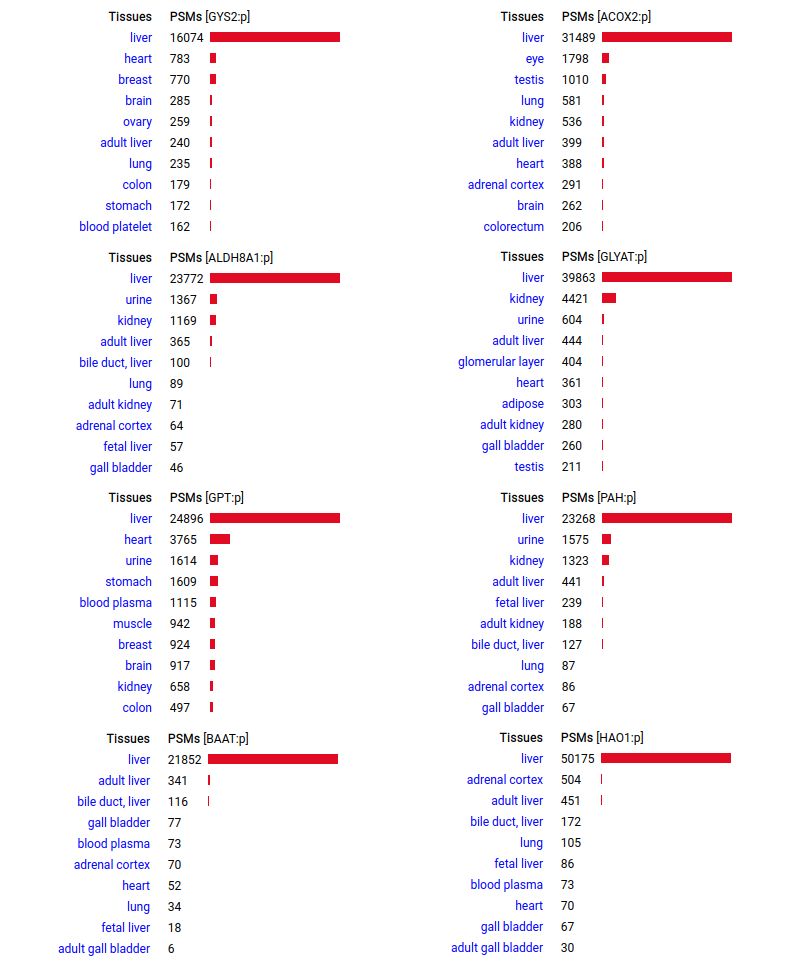
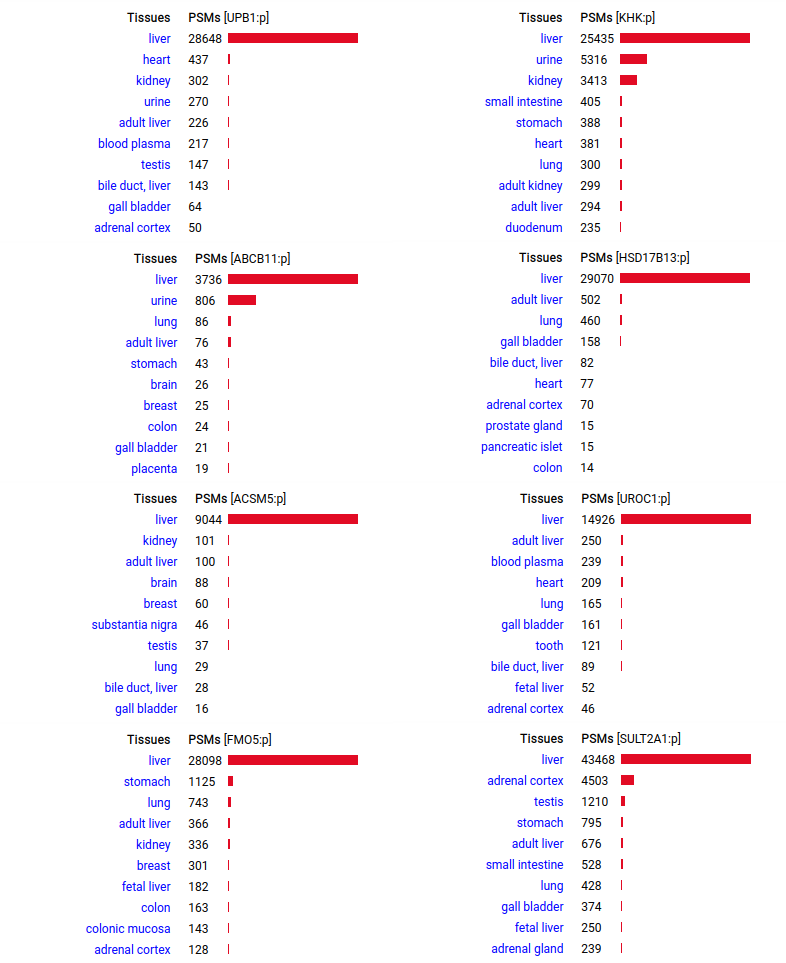
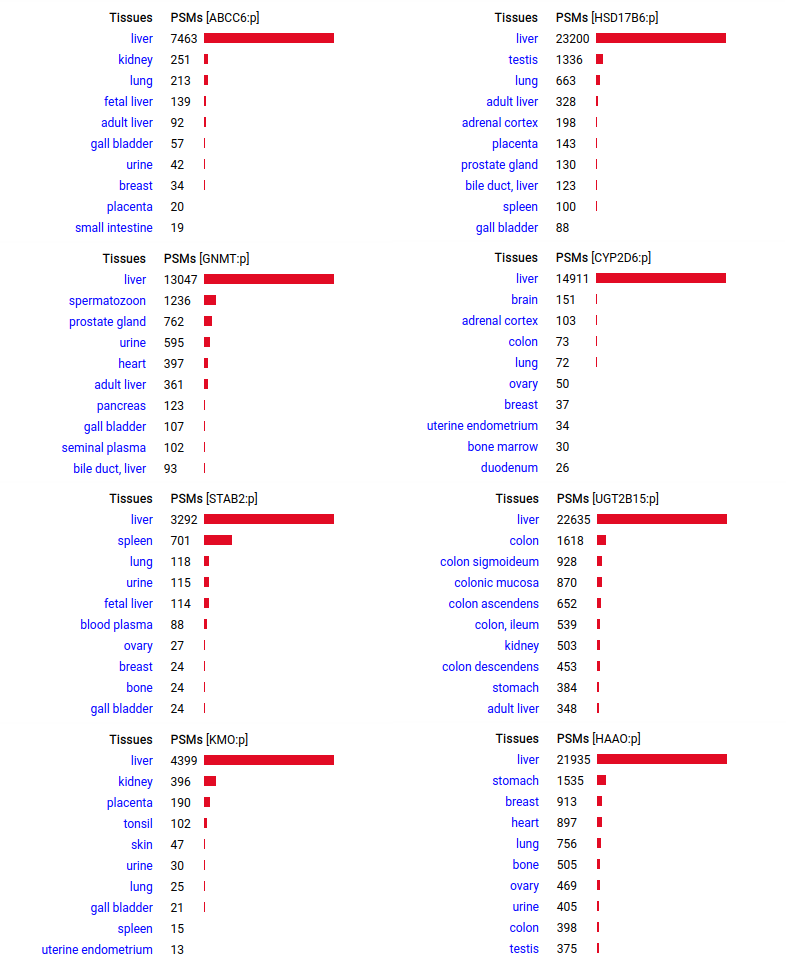
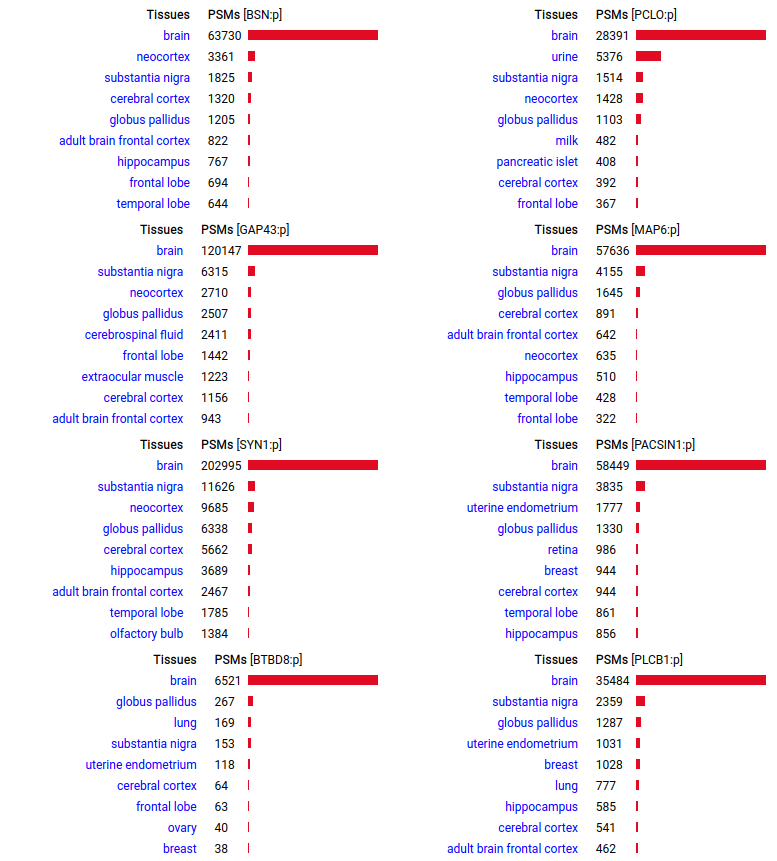
A scrapbook of select human brain-specific protein Q-stickers, that has been featured on Bluesky and Mastodon.
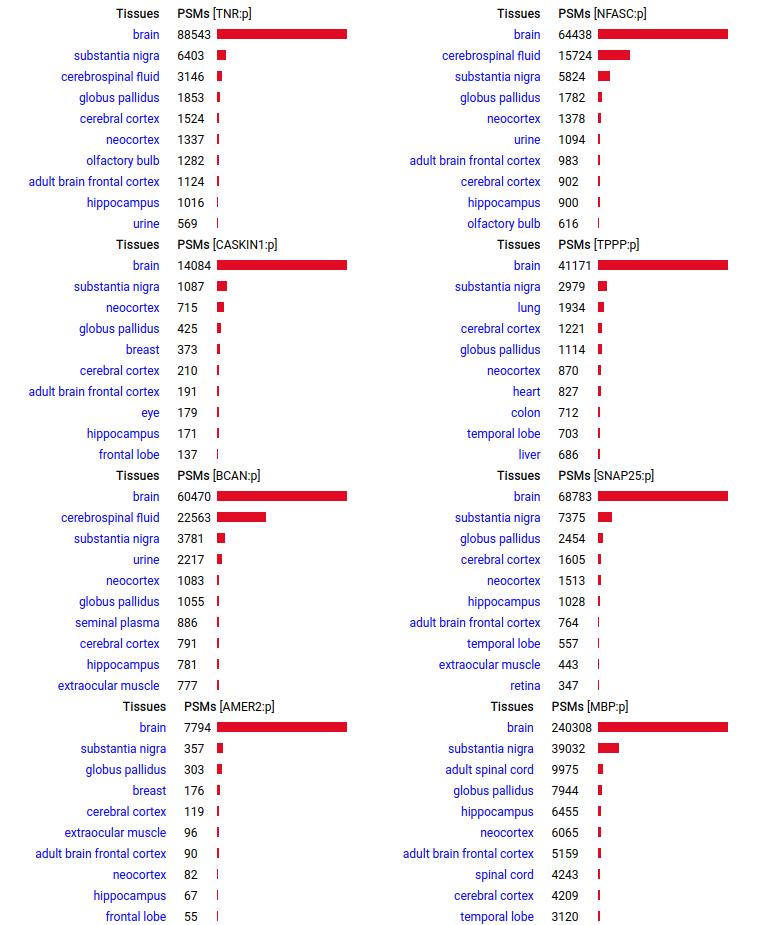
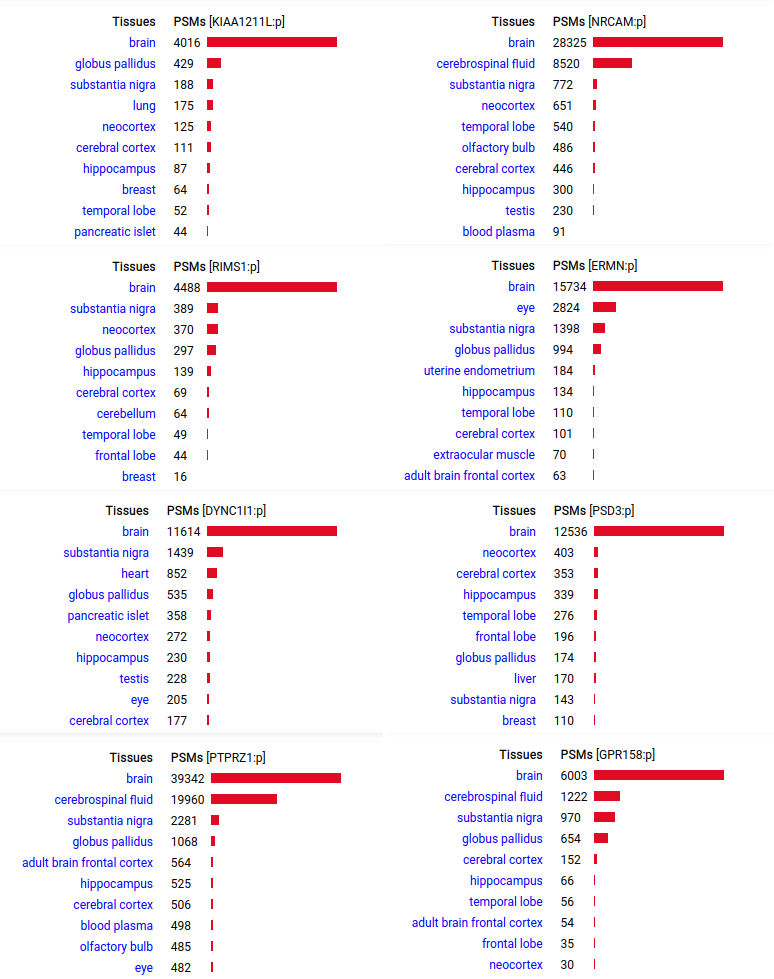
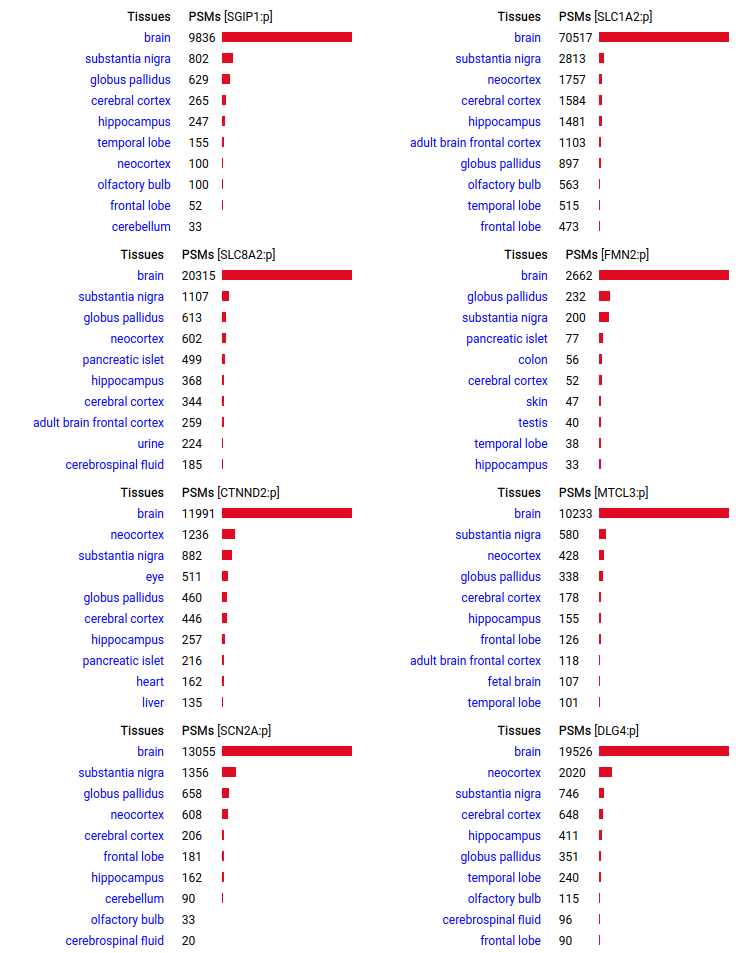
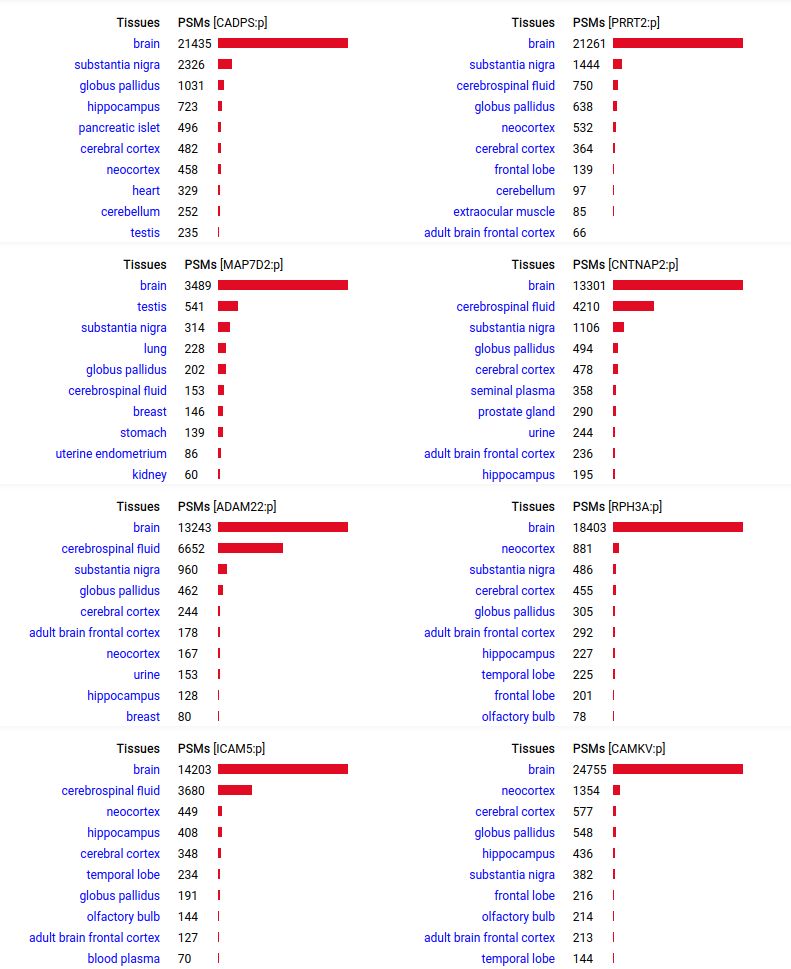
For several years, intrinsicdisorder.com has been used to display some diagrams associated with PTM and protein sequence structure.
Starting today, all references to intrinsicdisorder.com have been replaced by idr.thegpm.org in all URLs generated by GPMDB.
Because of the current trade war between the USA and Canada, US IP addresses are no longer allowed to make queries of GPMDB.
GPM Maintenence
(2024/07/028)
GPMDB and all associated interfaces will be off-line until 12:00 UTC 2024/07/29. This outage is necessary for the scheduled maintenance of
the system's hardware.
Assumptions: For each spectrum, follow these steps: Step 1: reduce the spectrum to a parent ion m/z and a set of peaks; Step 2: compare each spectrum to the candidate peptide sequences, using calculated b & y ions for the fragments & determine the number of calculated fragments that match the spectrum's peaks. Step 3: for the peptide sequence that provides the best match for a spectrum, calculate the probability (p) that a match is purely stochastic, using an appropriate statistical distribution (hypergeometric, Fisher exact test, simulation, etc). Step 4. Discard all PSMs where the calculated p is higher than an agreed upon significance test, e.g., p > 0.01. What remains are the PSMs you can use to assemble proteins or perform any sort of differential analysis. If you wish, at this point you can calculate the FDR, FPR or any other type of statistical test for the collection using the individual p values. You can do the same thing if you assume that each spectrum must be represented by a continuous function, but the math is messier. But, as you can tell, this sort of procedure has no brio, panache or mystery, reducing proteomics to being just like every other measurement that one might perform with a mass spectrometer. CanonFolly Everybody's canons select a sequence with 371 aa in 11 exons for human PRMT1:p (protein arginine methyltransferase 1). The proteomics data shows that assembly contains a ghost exon 2 (red highlight): the most commonly observed splice has 353 aa in 10 exons. 
The presence of the ghost exon affects the N-terminal phospho-IDR, shown here on the AlphaFold canonical sequence. 
CanonFolly ENSEMBL/REFSEQ/MANE canonize a sequence with 681 aa/6 exons for human ZFP64:p (zinc finger protein 64); UNIPROT/APRIS canonizes one with 645 aa/9 exons. The proteomics data shows they coexist, sharing their nward 5 exons (green), but the cward exons (red & blue) are not homologous. 
Canon Folly: the ENSEMBL/REFSEQ canonical transcript for human CPNE6:p (copine 6, 612 aa) doesn't seem to be initiating correctly, with a processed N-terminal resulting in S57+acetyl. An alternate transcript (557 aa) has this terminus at S2+acetyl & is a much better fit to proteomics experimental data.  
Canon folly: the ENSEMBL/REFSEQ/MANE canonical sequence of SPATA13:p (spermatogenesis associated 13, 1277 aa) disagrees with the UNIPROT selection (652 aa). The proteomics data shows that in most tissue the longer splice is present, but in T cells & macrophages it is the shorter splice that prevails. Protein-protein interaction expts have tended to use the 652 aa splice as bait.  
Canon folly: the UNIPROT canonical sequence for VPS53:p (vacuolar protein sorting 53 homolog subunit of the Golgi-associated retrograde protein complex 699 aa, 19 exons) disagrees with the ENSEMBL/MANE/REFSEQ canonical sequence (832 aa, 22 exons), which replaces 3' exon 19 & adds 20, 21 & 22. The proteomics data shows that in all tissues & cell lines the 22 exon splice is the norm. Protein-protein interaction expts have tended to use the bogus 19 exon splice as bait. 
Canon Folly: the canonical sequence chosen for COL18A1:p (human collagen type XVIII alpha 1) by UNIPROT/APRIS has 1754 aa in 41 exons. ENSEMBL/MANE/REFSEQ chose a 1339 aa contender with 42 exons in which a longish exon 1 (451 aa) is substituted for 2 short exons (36 aa total). You be the judge:  
Amusingly to me: in the mouse case, ENSEMBL/APRIS chose the long splice (1774 aa), even though the shorter splice (1315 aa) is again a much better fit to how experiments describe reality. Consistency is never a issue in biocuration: the only standard is a dedication to what ever "is written".  
Canon folly: MT1E:p (human metallothionein 1E) has an ENSEMBL/MANE/REFSEQ canonical form (127 aa, 2 exons) that is longer than the UNIPROT/APRIS canonical form (61 aa, 3 exons). They share 30 residues of sequence at the N-terminus. There is no evidence in proteomics data that the 127 aa form is present any tissue or cell line, but lots that the 61 aa form is broadly translated.  
Canon folly: CNS tissue proteomics data suggests that the version of SYNJ1:p canonized by UNIPROT (1573 aa, 31 exons) is missing its 368 residue C-terminal exon. The MANE/ENSEMBL/REFSEQ canons select a splice (1311 aa, 31 exons) that swaps out the last exon for a short 6 residue region that is found in CNS tissue.  
But the fun isn't done. Ectopic translation of SYNJ1:p is often found in cell lines & tumours in non-CNS tissue that seems to favour the 1573 aa splice, with many observations of peptides from the long version of exon 31. 
Canon Folly: MANE, APRIS, ENSEMBL & UNIPROT all agree that the canonical sequence of human ANK2:p is a big one: 3957 aa in 46 exons. Unfortunately, unless you study brain/CNS tissue, you may never see that one. Instead, you will see splices missing the 2085 residue mega-exon 38, producing what looks like (and is) a big gap in proteomics data coverage when analyzed using canon.    
Even though ANK2:p exon 38 only contributes signals in CNS/brain tissue samples, it still shows up nicely in overall averages and has some pretty active S/T phosphorylation acceptors. Remember this when reading a naive database blurb about a protein that casually mentions that there may be "isoforms".  
Canon Folly: the MANE/ENSEMBL/REFSEQ/APRIS canonized sequence for human CLIC5:p has 251 aa in 6 exons (CLIC5-A), while the UNIPROT canonized sequence has 410 aa in 6 exons (CLIC5-B): CLIC5-B has a long exon 1 replacing a short one in CLIC5-A. Proteomics data from almost all tissues & fluids are pretty conclusive: the 410 aa sequence is not detectable.    
But, there is a wrinkle. The data below shows CLIC5:p observations from PXD010154, a study that generated survey data in multiple tissues. The top 2 experiments (small intestine & duodenum) clearly demonstrate the existence of the 410 aa sequence in those tissues & 251 aa sequence in all others. 
Only the colored codons/residues are allowed at the N-terminus of a nascent protein sequence, as it emerges from a ribosomal exit tunnel. A blue residue at the N-terminus is normally N-acetylated, a green residue may be N-acetylated (but often isn't) & an orange residue is not N-acetylated. Exception: any protein with a sequence that starts with "XP" is not N-acetylated for X = S, T, A, C, V or G. 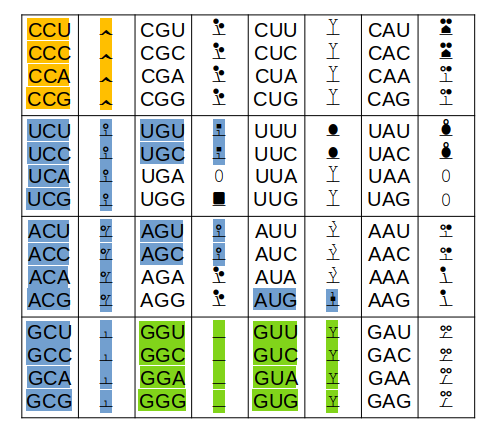
Other exceptions: 1 MR... will not be N-acetylated (tubulin exception) 1 MI... with not be N-acetylated (steric exception) 1 acetyl-MDDD (actin B exception) will mature to 1 acetyl-MEEE (actin G1 exception) will mature to The actin PTMs occur in the cytoplasm via enzymes dedicated to the purpose.
Something that has shaped the practice of proteomics was to base
as much as possible on sets of rules rather than statistics or other types
of evidence derived from the data itself.
The most fundamental of
these rules are the "canons" (fancy rules) used to select a single splice
variant to stand in for all of the possible translations of a gene. A particularly
influential set of 5 rules for this choice is used by UniProt
(see here).
It should come as no surprise that these rather trivial rules regularly choose a
splice that isn't very representative of what is found in biological samples, which can cause
bias and systematic interpretation errors in the results of typical data interpretation methods.
The fumarase gene is responsible for both a mitochondrial & cytoplasmic form that differ only in that the cytoplasmic form has an N-terminal acetylation. When the translation of FH:r initiates at its first Met codon ... ACC AUG UAC ... the sequence has a 44 residue mitochondrial transit peptide that is removed on the passage of the sequence into the mitochondrial inner matrix. Human fumarate hydratase (FH:p) 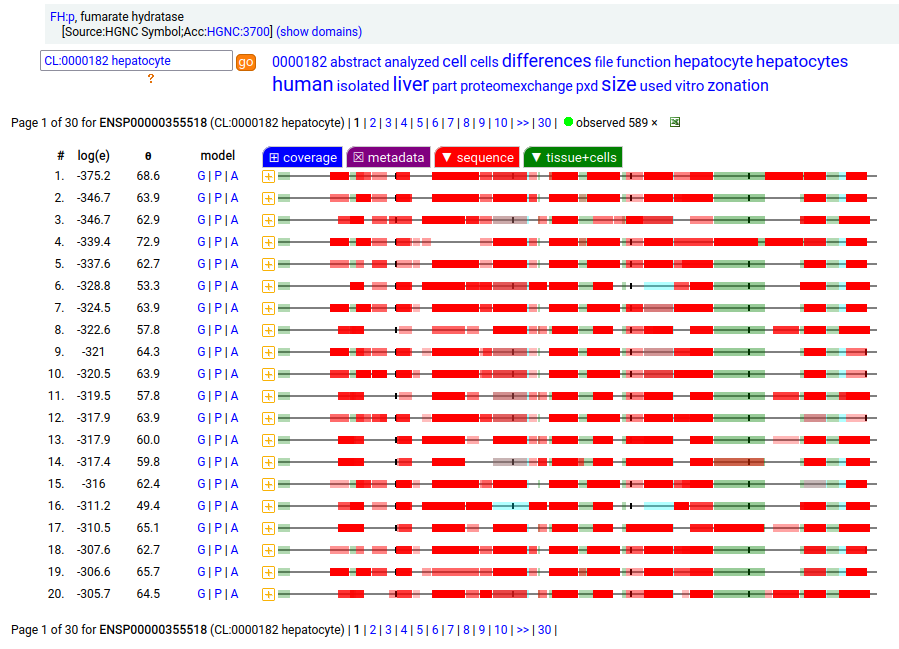
It seems that about 10% translations skip this first Met-codon & instead initiate at the second one ... CGA AUG GCA ... resulting in a protein with the N-terminal tryptic peptides 45 ac-ASQNSFR 51 lacking the transit peptide, so it stays in the cytoplasm. The gene pulls off this trick by having the second Met codon as the last codon of the first exon, which also codes for the entire transit peptide. 
MTCH1:p is interesting: it has 2 initiation variants that are observed to be tissue-dependent. Brain tissue has the M1 initiating variant as the dominant form of this membrane protein, while liver tissue has the M19 initiating variant as the dominant form. Human mitochondrial carrier 1 (MTCH1:p) proteomics canon_folly 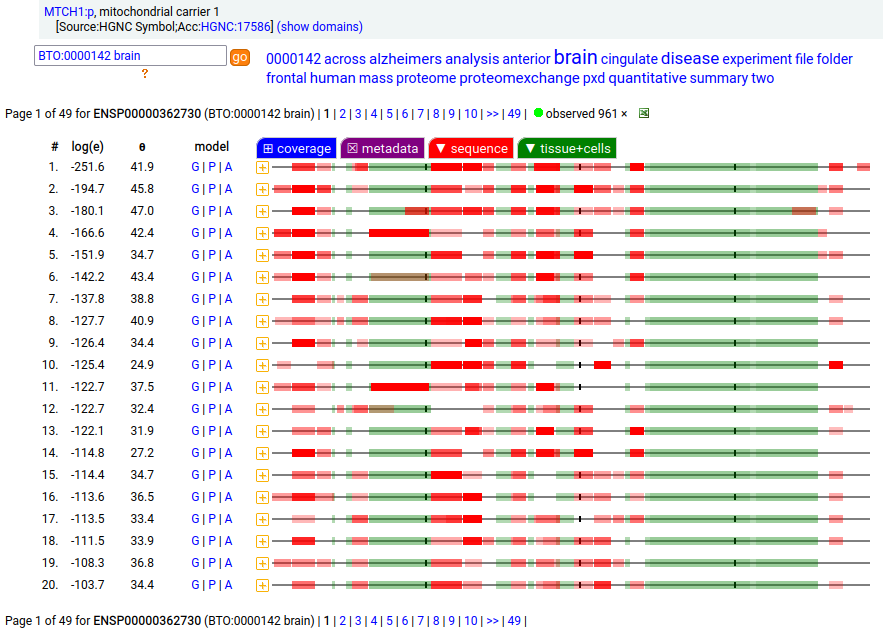 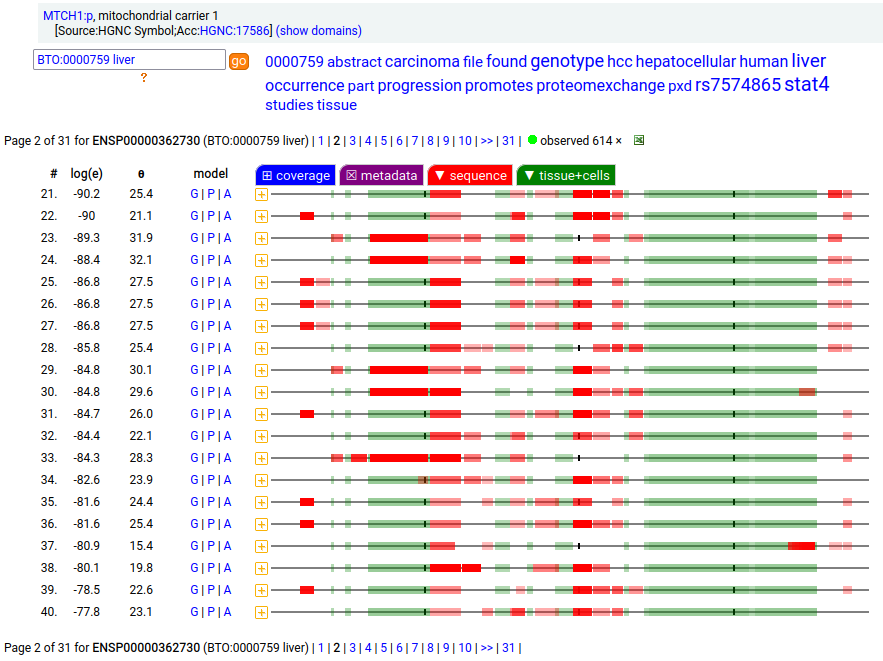
This sequence also has overlapping tryptic peptides that can be used to monitor the amount of each variant present in a sample. Mouse appears to use the same mechanism & shares the same tissue variant distribution. 
MTCH2:r has an AUG at about the same position in its mRNA sequence as MTCH1:r, but it does not generate observable levels of an initiation variant protein product. The coverage diagram for the canonical (252 aa) form of DUT:p may look like there is a splice selection error, but there is something more subtle going on. In fact, this diagram is characteristic of the removal of a longish mitochrondrial targeting peptide (1-69,70), resulting in a shorter mature enzyme in the mitochondrial matrix with a slightly ragged N-terminus. Human deoxyuridine triphosphatase (DUT:p) 
But, DUT:p is needed in both the mitochondria & cytoplasm. Rather than having a 2nd gene, in this case DUT:p has another splice variant (162 aa) that effectively removes the targeting peptide but retains the enzymatic portion of the sequence by removing the 2 exons coding for the peptide in the longer splice & substituting a shorter exon with an AUG in the right place. 
These two splices can be distinguished in proteomics data by examining the mature form N-terminal tryptic peptides corresponding to the two different protein sequences. Note: N-terminal proline residues do not undergo co-translationally acetylation. 
GARS1:p is another case where the coverage diagram suggests there may be some RNA high jinks at the N-terminus. The full length (739 aa) translation has a mitochondrial transit peptide (1-35,37,39) that is removed during import into the mitochondrial matrix, leaving the enzyme with a ragged N-terminus. Human glycyl-tRNA synthetase 1 (GARS1:p) 
To create the more abundant cytoplasmic protein, neither a new gene nor splice is necessary. Instead, it is generated by simply ignoring M1 during translation, skipping the transit peptide and initiating at M55 (685 aa). While any tryptic peptide with a residues in the region (35-54) must be from the mitochondrial enzyme, only peptides with acetylated M55 at their N-terminus are unambiguously cytoplasmic. 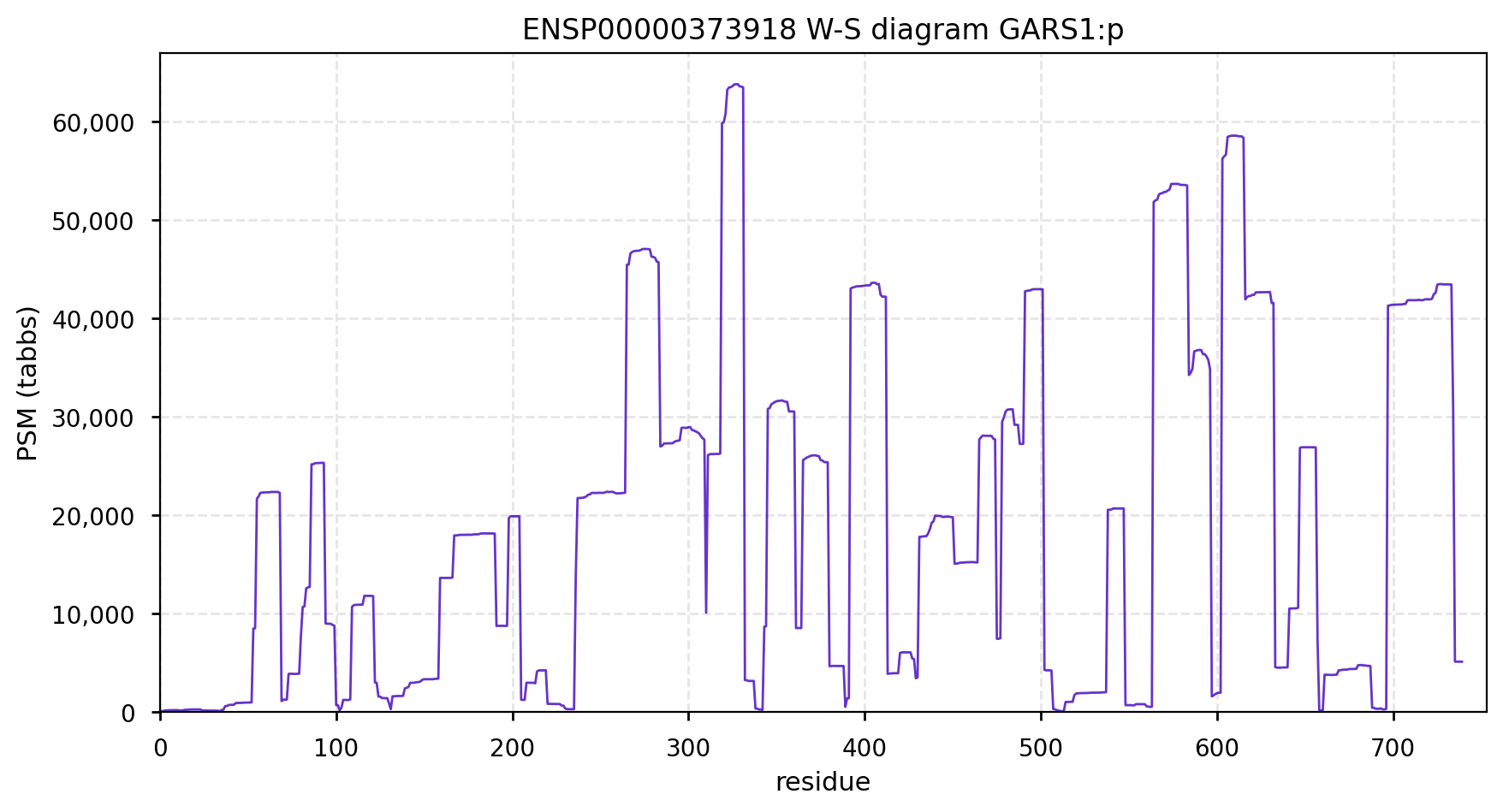 
Canon Folly: Palladin's ENSEMBL/MANE/INSDC/RefSeq canonical sequence (1223 aa) isn't detectable in vivo, while a shorter splice (777 aa) is widely distributed. In the images below, the observed coverage diagrams show the same 20 LC/MS/MS runs, fit to either the canonical or (777 aa) sequence. The 2 on top are bait-prey protein-protein interaction experiments with a recombinant canonical sequence as the bait. Human palladin, cytoskeletal associated protein (PALLD:p)  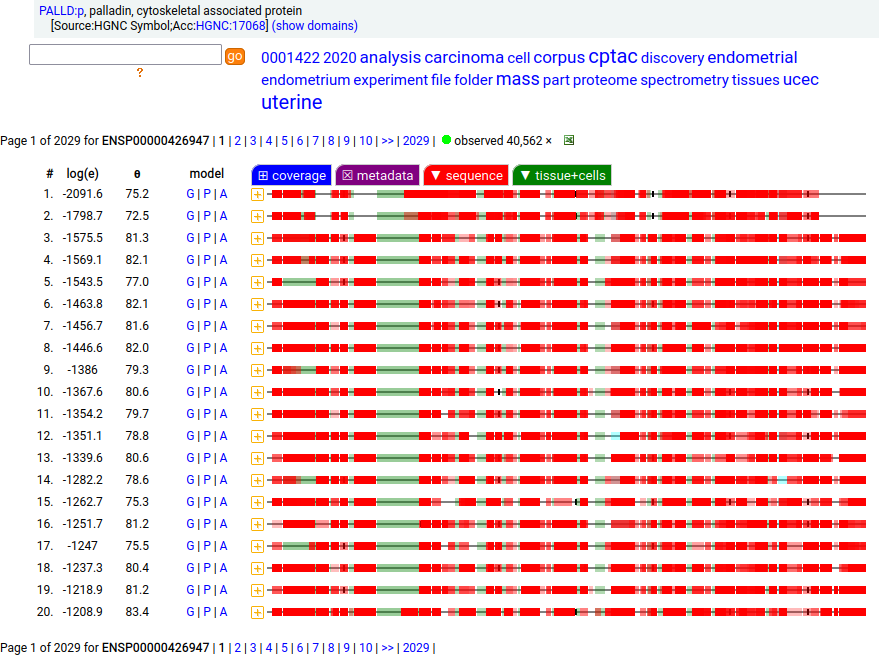
There is another splice variant (1106 aa) that appears to only be expressed in striated muscle (skeletal & cardiac). The (777 aa) form is used in smooth muscle (e.g., blood vessels, urinary bladder, colon). 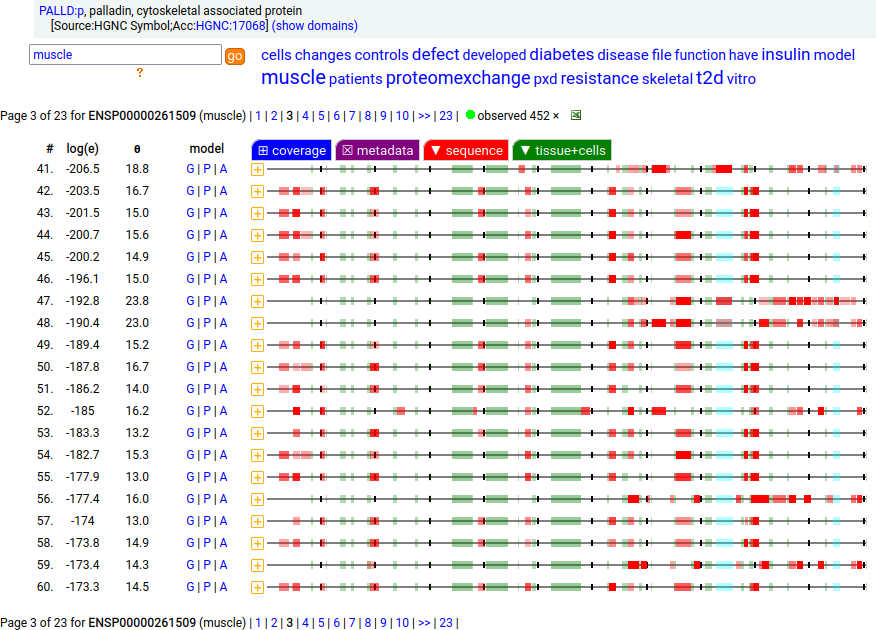
Canon Folly: FAM98B:p has a UP/ENSEMBL/MANE/CCDS/REFSEQ canonical sequence (433 aa) that is observed initiating at M1 (not acetylated), but a shorter version (422 aa) that initiates at M12 is acetylated and observed just as often in the same samples. The 3 peptides shown are useful to determine which initiation variants are present. It appears mammals all use this particular trick. Human family with sequence similarity 98, member B (FAM98B:p) 
Canon Folly: UniProt/ENSEMBL/MANE/CCDS/INSDC all agree that PNPO:g should result in the same canonical protein sequence (261 aa). Unfortunately the predominant observed protein (230 aa) is the result of translation initiation & N-acetylation at M32, the 2nd AUG in-frame on the mRNA. Human pyridoxamine 5'-phosphate oxidase (PNPO:p) 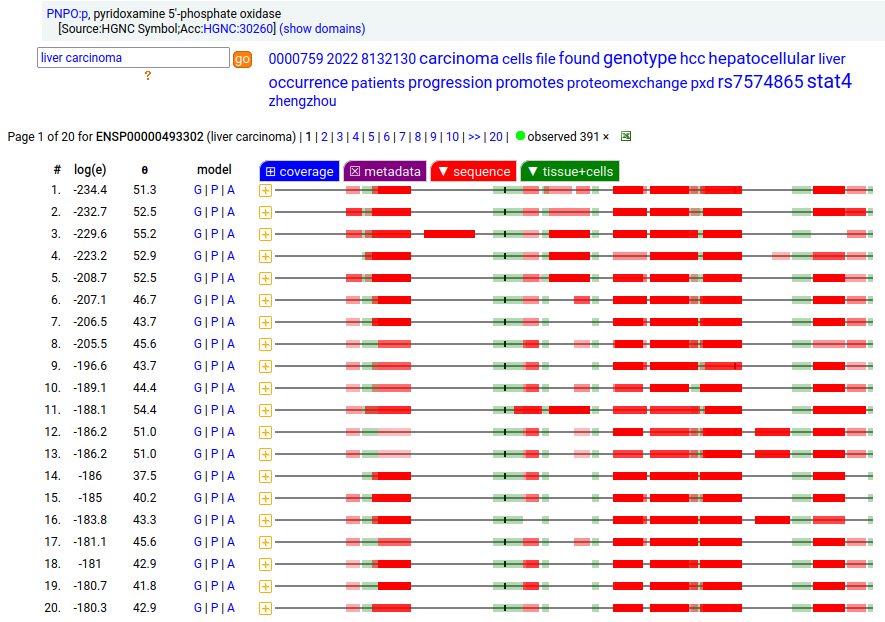
Canon Folly: Disagreements between canons is not unusual. REPIN1:p has two proposed canonical forms: ENSEMBL's with 2 exons (624 aa) and UNIPROT's 1 exon form (567 aa). The data has no observations of the peptides in the 1st exon of the 624 aa form , suggesting very strongly that the dominant translated "proteoform" is the 1 exon sequence. Human replication initiator 1 (REPIN1:p) 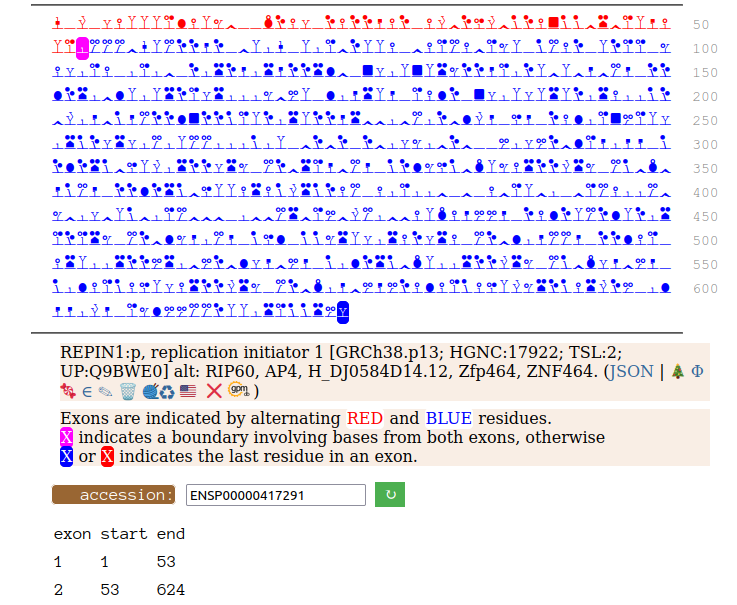 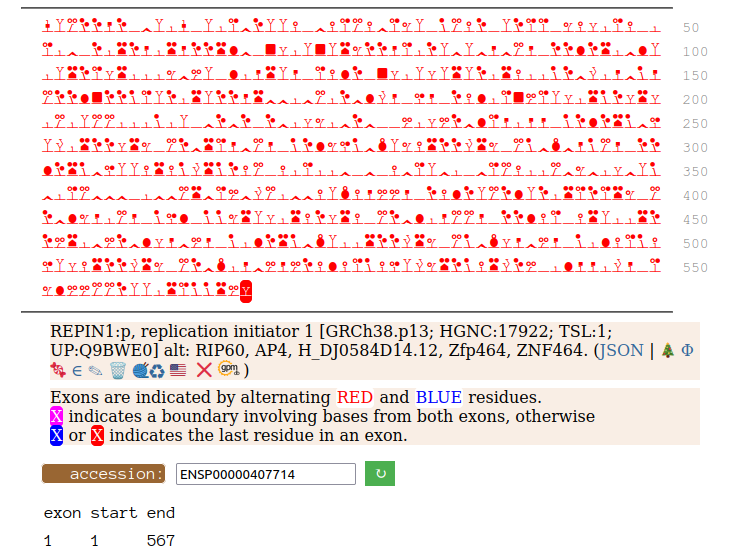
Canon Folly: NDUFV3:p is part of mitochondrial complex I. Its UP canonical sequence (108 aa) may be observed, but a different splice with an additional exon (473 aa) is much more common. Shown below are the coverage patterns observed for larger splice in BioPlex Interactome project data, with & without the UP canonical sequence as a bait. Human NADH:ubiquinone oxidoreductase subunit V3 (NDUFV3:p) 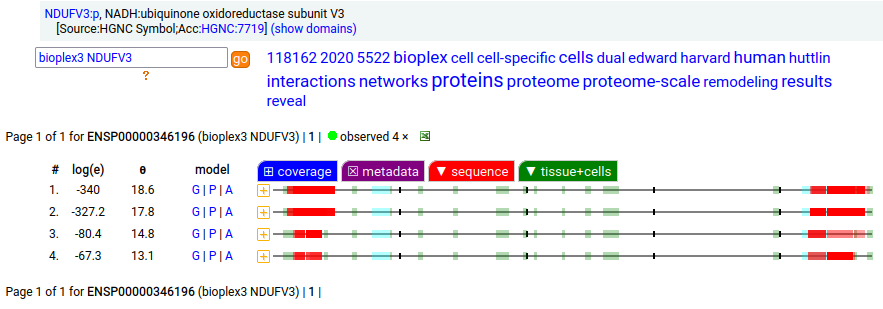 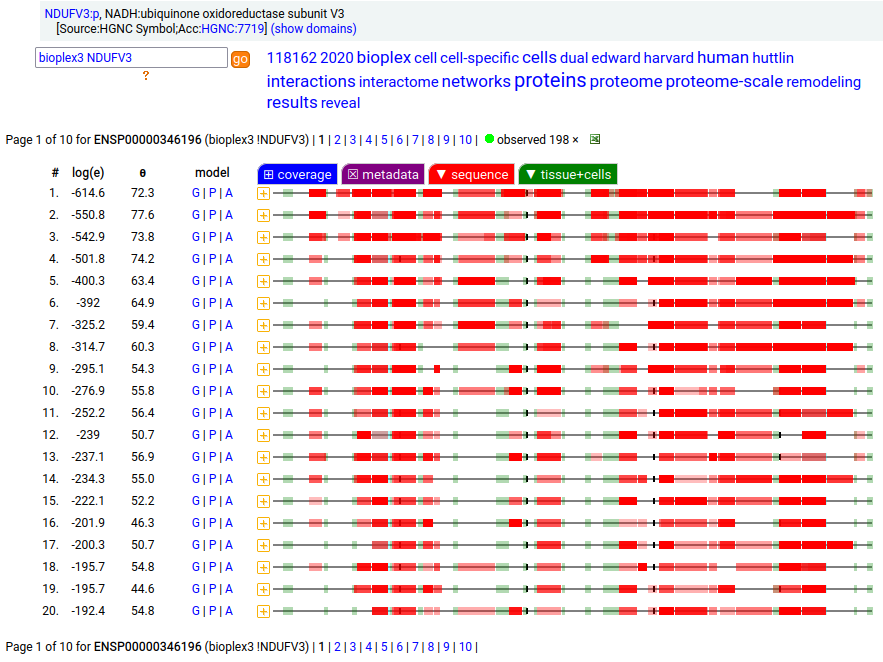
On the off chance that someone may be interested, here are the sequences of the 2 splices, with the exons marked.  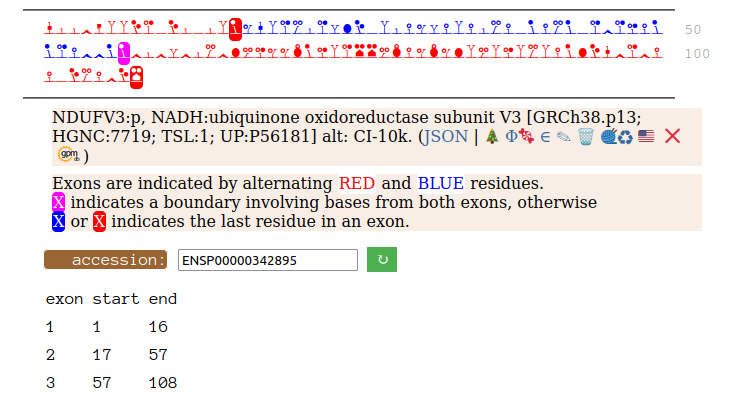
Canon Folly: ENSEMBL & UP canonical TSC22D1:p (1073 aa) is observed, but so is a much smaller splice (144 aa), which initiates at its M11. Both splices share 2 C-terminal exons, swapping out a single, long N-terminal exon (970 aa) for a short one (42 aa). BioPlex3 project data shows these 2 variants have different protein-protein interaction partners. Human transcription factor transforming growth factor β1-stimulated clone 22 domain family member 1 (TSC22D1:p) Canon Folly: The "canonical" sequence of UBAP2:p (1119 aa) is rarely translated, in favour of a subtly different, slightly shorter version (1117 aa). M1: canonical initiation site (1:20), co-translationally acetylated when this occurs Human ubiquitin associated protein 2 (UBAP2:p) Canon Folly: The "canonical" sequence (510 aa) of SYT6:p simply doesn't seem to exist in nature. Another GENCODE splice variant (425 aa) is closer, but the dominant sequence in tissue initiates at M30 rather than M1 of this splice (395 aa). The 425 aa sequence is seen occasionally as the bait in protein-protein interaction studies. Human synaptotagmin VI (SYT6:p)―GPMDB peptide identification diagram. 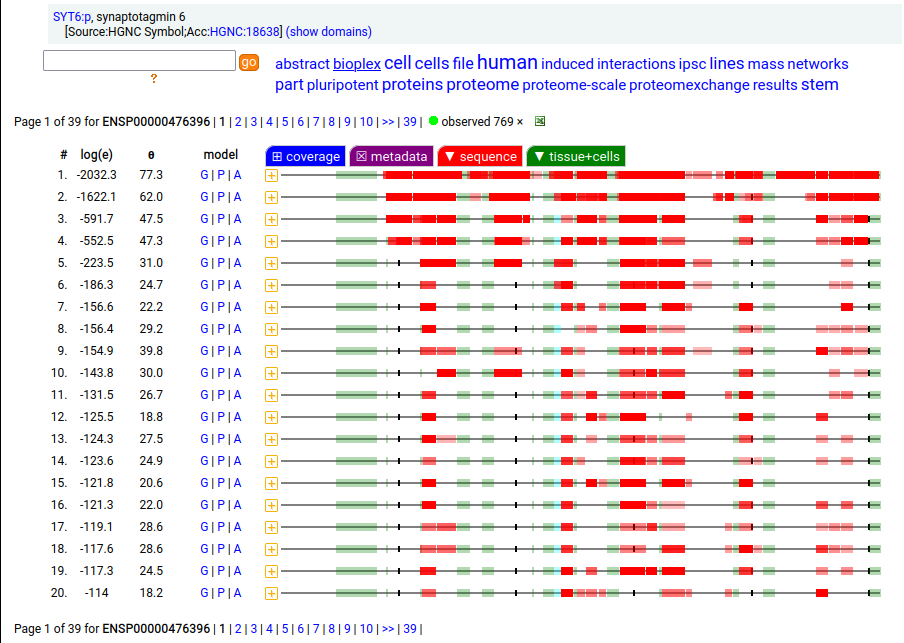 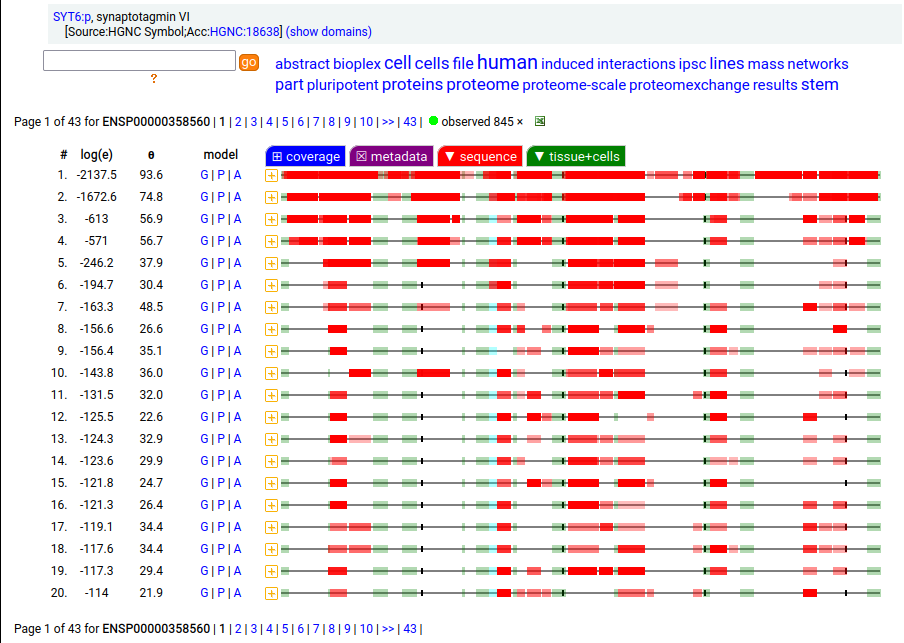
Canon Folly: which splice is the "right" splice? For the young, LEPR:p was one of the hottest targets in drug development for quite a while, but was also the ruin of many a scientist―it's ligand may have been the last nail in the coffin of the "protein engineering" craze back in the late 80's, early 90's.  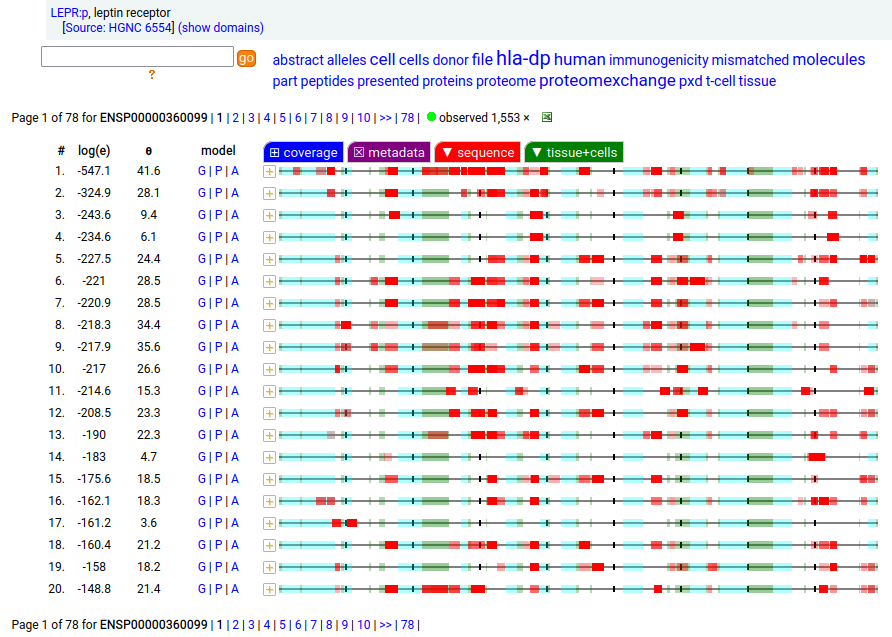
Copyright © 2024, The Global Proteome Machine.
Located at 137 Bannatyne | Privacy Statement
|